bedtools: a powerful toolset for genome arithmetic¶
Collectively, the bedtools utilities are a swiss-army knife of tools for a wide-range of genomics analysis tasks. The most widely-used tools enable genome arithmetic: that is, set theory on the genome. For example, bedtools allows one to intersect, merge, count, complement, and shuffle genomic intervals from multiple files in widely-used genomic file formats such as BAM, BED, GFF/GTF, VCF. While each individual tool is designed to do a relatively simple task (e.g., intersect two interval files), quite sophisticated analyses can be conducted by combining multiple bedtools operations on the UNIX command line.
bedtools is developed in the Quinlan laboratory at the University of Utah and benefits from fantastic contributions made by scientists worldwide.
Tutorial¶
- We have developed a fairly comprehensive tutorial that demonstrates both the basics, as well as some more advanced examples of how bedtools can help you in your research. Please have a look.
- Robert Aboukhalil has developed sandbox.bio an excellent, web-based playground for the bedtools tutorial and other widely-used genomics tools.
Important notes¶
- As of version 2.28.0, bedtools now supports the CRAM format via the use of htslib. Specify the reference genome associated with your CRAM file via the CRAM_REFERENCE environment variable. Bedtools will look for this environment variable when it needs to access sequence data from the CRAM file (e.g., bamtofastq).
- With the exception of BAM files, bedtools assumes all input files are TAB delimited.
- bedtools also assumes that all input files use UNIX line endings.
- Unless you use the -sorted option, bedtools currently does not support chromosomes larger than 512Mb
- When using the -sorted option with files whose chromosomes are not lexicographically sorted (e.g., sort -k1,1 -k2,2n for BED files), one must provide a genome file (-g) defining the expected chromosome order.
- bedtools requires that chromosome naming schemes are identical in files that you are comparing (e.g., ‘chr1’ in one file and ‘1’ in another won’t work).
- .fai files may be used as genome (-g) files.
Interesting Usage Examples¶
In addition, here are a few examples of how bedtools has been used for genome research. If you have interesting examples, please send them our way and we will add them to the list.
- Coverage analysis for targeted DNA capture. Thanks to Stephen Turner.
- Measuring similarity of DNase hypersensitivity among many cell types
- Extracting promoter sequences from a genome
- Comparing intersections among many genome interval files
- RNA-seq coverage analysis. Thanks to Erik Minikel.
- Identifying targeted regions that lack coverage. Thanks to Brent Pedersen.
- Calculating GC content for CCDS exons.
- Making a master table of ChromHMM tracks for multiple cell types.
Table of contents¶
Overview¶
Background¶
The development of bedtools was motivated by a need for fast, flexible tools with which to compare large sets of genomic features. Answering fundamental research questions with existing tools was either too slow or required modifications to the way they reported or computed their results. We were aware of the utilities on the UCSC Genome Browser and Galaxy websites, as well as the elegant tools available as part of Jim Kent’s monolithic suite of tools (“Kent source”). However, we found that the web-based tools were too cumbersome when working with large datasets generated by current sequencing technologies. Similarly, we found that the Kent source command line tools often required a local installation of the UCSC Genome Browser. These limitations, combined with the fact that we often wanted an extra option here or there that wasn’t available with existing tools, led us to develop our own from scratch. The initial version of bedtools was publicly released in the spring of 2009. The current version has evolved from our research experiences and those of the scientists using the suite over the last year. The bedtools suite enables one to answer common questions of genomic data in a fast and reliable manner. The fact that almost all the utilities accept input from “stdin” allows one to “stream / pipe” several commands together to facilitate more complicated analyses. Also, the tools allow fine control over how output is reported. The initial version of bedtools supported solely 6-column BED files. However, we have subsequently added support for sequence alignments in BAM format, as well as for features in GFF , “blocked” BED format, and VCF format. The tools are quite fast and typically finish in a matter of a few seconds, even for large datasets. This manual seeks to describe the behavior and available functionality for each bedtool. Usage examples are scattered throughout the text, and formal examples are provided in the last two sections, we hope that this document will give you a sense of the flexibility of the toolkit and the types of analyses that are possible with bedtools. If you have further questions, please join the bedtools discussion group, visit the Usage Examples on the Google Code site (usage, advanced usage), or take a look at the nascent “Usage From the Wild” page.
Summary of available tools.¶
bedtools support a wide range of operations for interrogating and manipulating genomic features. The table below summarizes the tools available in the suite.
| Utility | Description |
|---|---|
| annotate | Annotate coverage of features from multiple files. |
| bamtobed | Convert BAM alignments to BED (& other) formats. |
| bamtofastq | Convert BAM records to FASTQ records. |
| bed12tobed6 | Breaks BED12 intervals into discrete BED6 intervals. |
| bedpetobam | Convert BEDPE intervals to BAM records. |
| bedtobam | Convert intervals to BAM records. |
| closest | Find the closest, potentially non-overlapping interval. |
| cluster | Cluster (but don’t merge) overlapping/nearby intervals. |
| complement | Extract intervals _not_ represented by an interval file. |
| coverage | Compute the coverage over defined intervals. |
| expand | Replicate lines based on lists of values in columns. |
| fisher | Calculate Fisher statistic b/w two feature files. |
| flank | Create new intervals from the flanks of existing intervals. |
| genomecov | Compute the coverage over an entire genome. |
| getfasta | Use intervals to extract sequences from a FASTA file. |
| groupby | Group by common cols. & summarize oth. cols. (~ SQL “groupBy”) |
| igv | Create an IGV snapshot batch script. |
| intersect | Find overlapping intervals in various ways. |
| jaccard | Calculate the Jaccard statistic b/w two sets of intervals. |
| links | Create a HTML page of links to UCSC locations. |
| makewindows | Make interval “windows” across a genome. |
| map | Apply a function to a column for each overlapping interval. |
| maskfasta | Use intervals to mask sequences from a FASTA file. |
| merge | Combine overlapping/nearby intervals into a single interval. |
| multicov | Counts coverage from multiple BAMs at specific intervals. |
| multiinter | Identifies common intervals among multiple interval files. |
| nuc | Profile the nucleotide content of intervals in a FASTA file. |
| overlap | Computes the amount of overlap from two intervals. |
| pairtobed | Find pairs that overlap intervals in various ways. |
| pairtopair | Find pairs that overlap other pairs in various ways. |
| random | Generate random intervals in a genome. |
| reldist | Calculate the distribution of relative distances b/w two files. |
| tools/sample | Sample random records from file using reservoir sampling. |
| shift | Adjust the position of intervals. |
| shuffle | Randomly redistribute intervals in a genome. |
| slop | Adjust the size of intervals. |
| sort | Order the intervals in a file. |
| tools/spacing | Sample random records from file using reservoir sampling. |
| tools/split | Split a file into multiple files with equal records or base pairs. |
| subtract | Remove intervals based on overlaps b/w two files. |
| tag | Tag BAM alignments based on overlaps with interval files. |
| unionbedg | Combines coverage intervals from multiple BEDGRAPH files. |
| window | Find overlapping intervals within a window around an interval. |
Fundamental concepts.¶
What are genome features and how are they represented?¶
Throughout this manual, we will discuss how to use bedtools to manipulate, compare and ask questions of genome “features”. Genome features can be functional elements (e.g., genes), genetic polymorphisms (e.g. SNPs, INDELs, or structural variants), or other annotations that have been discovered or curated by genome sequencing groups or genome browser groups. In addition, genome features can be custom annotations that an individual lab or researcher defines (e.g., my novel gene or variant).
The basic characteristics of a genome feature are the chromosome or scaffold on which the feature “resides”, the base pair on which the feature starts (i.e. the “start”), the base pair on which feature ends (i.e. the “end”), the strand on which the feature exists (i.e. “+” or “-“), and the name of the feature if one is applicable.
The two most widely used formats for representing genome features are the BED (Browser Extensible Data) and GFF (General Feature Format) formats. bedtools was originally written to work exclusively with genome features described using the BED format, but it has been recently extended to seamlessly work with BED, GFF and VCF files.
Existing annotations for the genomes of many species can be easily downloaded in BED and GFF format from the UCSC Genome Browser’s “Table Browser” (http://genome.ucsc.edu/cgi-bin/hgTables?command=start) or from the “Bulk Downloads” page (http://hgdownload.cse.ucsc.edu/downloads.html). In addition, the Ensemble Genome Browser contains annotations in GFF/GTF format for many species (http://www.ensembl.org/info/data/ftp/index.html)
Overlapping / intersecting features.¶
Two genome features (henceforth referred to as “features”) are said to overlap or intersect if they share at least one base in common. In the figure below, Feature A intersects/overlaps Feature B, but it does not intersect/overlap Feature C.
TODO: place figure here
Comparing features in file “A” and file “B”.¶
The previous section briefly introduced a fundamental naming convention used in bedtools. Specifically, all bedtools that compare features contained in two distinct files refer to one file as feature set “A” and the other file as feature set “B”. This is mainly in the interest of brevity, but it also has its roots in set theory. As an example, if one wanted to look for SNPs (file A) that overlap with exons (file B), one would use bedtools intersect in the following manner:
bedtools intersect –a snps.bed –b exons.bed
There are two exceptions to this rule: 1) When the “A” file is in BAM format, the “-abam” option must be used. For example:
bedtools intersect –abam alignedReads.bam –b exons.bed
And 2) For tools where only one input feature file is needed, the “-i” option is used. For example:
bedtools merge –i repeats.bed
BED starts are zero-based and BED ends are one-based.¶
bedtools users are sometimes confused by the way the start and end of BED features are represented. Specifically, bedtools uses the UCSC Genome Browser’s internal database convention of making the start position 0-based and the end position 1-based: (http://genome.ucsc.edu/FAQ/FAQtracks#tracks1) In other words, bedtools interprets the “start” column as being 1 basepair higher than what is represented in the file. For example, the following BED feature represents a single base on chromosome 1; namely, the 1st base:
chr1 0 1 first_base
Why, you might ask? The advantage of storing features this way is that when computing the length of a feature, one must simply subtract the start from the end. Were the start position 1-based, the calculation would be (slightly) more complex (i.e. (end-start)+1). Thus, storing BED features this way reduces the computational burden.
GFF starts and ends are one-based.¶
In contrast, the GFF format uses 1-based coordinates for both the start and the end positions. bedtools is aware of this and adjusts the positions accordingly. In other words, you don’t need to subtract 1 from the start positions of your GFF features for them to work correctly with bedtools.
VCF coordinates are one-based.¶
The VCF format uses 1-based coordinates. As in GFF, bedtools is aware of this and adjusts the positions accordingly. In other words, you don’t need to subtract 1 from the start positions of your VCF features for them to work correctly with bedtools.
File B is loaded into memory (most of the time).¶
Whenever a bedtool compares two files of features, the “B” file is loaded into memory. By contrast, the “A” file is processed line by line and compared with the features from B. Therefore to minimize memory usage, one should set the smaller of the two files as the B file. One salient example is the comparison of aligned sequence reads from a current DNA sequencer to gene annotations. In this case, the aligned sequence file (in BED format) may have tens of millions of features (the sequence alignments), while the gene annotation file will have tens of thousands of features. In this case, it is wise to sets the reads as file A and the genes as file B.
Feature files must be tab-delimited.¶
This is rather self-explanatory. While it is possible to allow BED files to be space-delimited, we have decided to require tab delimiters for three reasons:
- By requiring one delimiter type, the processing time is minimized.
- Tab-delimited files are more amenable to other UNIX utilities.
- GFF files can contain spaces within attribute columns. This complicates the use of space-delimited files as spaces must therefore be treated specially depending on the context.
All bedtools allow features to be “piped” via standard input.¶
In an effort to allow one to combine multiple bedtools and other UNIX utilities into more complicated “pipelines”, all bedtools allow features to be passed to them via standard input. Only one feature file may be passed to a bedtool via standard input. The convention used by all bedtools is to set either file A or file B to “stdin” or “-”. For example:
cat snps.bed | bedtools intersect –a stdin –b exons.bed
cat snps.bed | bedtools intersect –a - –b exons.bed
In addition, all bedtools that simply require one main input file (the -i file) will assume that input is coming from standard input if the -i parameter is ignored. For example, the following are equivalent:
cat snps.bed | bedtools sort –i stdin
cat snps.bed | bedtools sort
Most bedtools write their results to standard output.¶
To allow one to combine multiple bedtools and other UNIX utilities into more complicated “pipelines”, most bedtools report their output to standard output, rather than to a named file. If one wants to write the output to a named file, one can use the UNIX “file redirection” symbol “>” to do so. Writing to standard output (the default):
bedtools intersect –a snps.bed –b exons.bed
chr1 100100 100101 rs233454
chr1 200100 200101 rs446788
chr1 300100 300101 rs645678
Writing to a file:
bedtools intersect –a snps.bed –b exons.bed > snps.in.exons.bed
cat snps.in.exons.bed
chr1 100100 100101 rs233454
chr1 200100 200101 rs446788
chr1 300100 300101 rs645678
What is a “genome” file?¶
Some of the bedtools (e.g., genomecov, complement, slop) need to know the size of
the chromosomes for the organism for which your BED files are based. When using the UCSC Genome
Browser, Ensemble, or Galaxy, you typically indicate which species / genome build you are working.
The way you do this for bedtools is to create a “genome” file, which simply lists the names of the
chromosomes (or scaffolds, etc.) and their size (in basepairs).
Genome files must be tab-delimited and are structured as follows (this is an example for C. elegans):
chrI 15072421
chrII 15279323
...
chrX 17718854
chrM 13794
bedtools includes predefined genome files for human and mouse in the /genomes directory included in the bedtools distribution. Additionally, the “chromInfo” files/tables available from the UCSC Genome Browser website are acceptable. For example, one can download the hg19 chromInfo file here: http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/chromInfo.txt.gz
Paired-end BED files (BEDPE files).¶
We have defined a new file format (BEDPE) to concisely describe disjoint genome features, such as structural variations or paired-end sequence alignments. We chose to define a new format because the existing BED block format (i.e. BED12) does not allow inter-chromosomal feature definitions. Moreover, the BED12 format feels rather bloated when one want to describe events with only two blocks.
Use “-h” for help with any bedtool.¶
Rather straightforward. If you use the “-h” option with any bedtool, a full menu of example usage and available options (when applicable) will be reported.
BED features must not contain negative positions.¶
bedtools will typically reject BED features that contain negative positions. In special cases, however, BEDPE positions may be set to -1 to indicate that one or more ends of a BEDPE feature is unaligned.
The start position must be <= to the end position.¶
bedtools will reject BED features where the start position is greater than the end position.
Headers are allowed in GFF and BED files¶
bedtools will ignore headers at the beginning of BED and GFF files. Valid header lines begin with a “#” symbol, the work “track”, or the word “browser”. For example, the following examples are valid headers for BED or GFF files:
track name=aligned_read description="Illumina aligned reads”
chr5 100000 500000 read1 50 +
chr5 2380000 2386000 read2 60 -
#This is a fascinating dataset
chr5 100000 500000 read1 50 +
chr5 2380000 2386000 read2 60 -
browser position chr22:1-20000
chr5 100000 500000 read1 50 +
chr5 2380000 2386000 read2 60 -
GZIP support: BED, GFF, VCF, and BEDPE file can be “gzipped”¶
bedtools will process gzipped BED, GFF, VCF and BEDPE files in the same manner as uncompressed files. Gzipped files are auto-detected thanks to a helpful contribution from Gordon Assaf.
Support for “split” or “spliced” BAM alignments and “blocked” BED features¶
As of Version 2.8.0, five bedtools (intersect, coverage, genomecov,
bamToBed, and bed12ToBed6) can properly handle “split”/”spliced” BAM alignments (i.e., having an
“N” CIGAR operation) and/or “blocked” BED (aka BED12) features.
intersect, coverage, and genomecov will optionally handle “split” BAM and/or
“blocked” BED by using the -split option. This will cause intersects or coverage to be computed only
for the alignment or feature blocks. In contrast, without this option, the intersects/coverage would be
computed for the entire “span” of the alignment or feature, regardless of the size of the gaps between
each alignment or feature block. For example, imagine you have a RNA-seq read that originates from
the junction of two exons that were spliced together in a mRNA. In the genome, these two exons
happen to be 30Kb apart. Thus, when the read is aligned to the reference genome, one portion of the
read will align to the first exon, while another portion of the read will align ca. 30Kb downstream to the
other exon. The corresponding CIGAR string would be something like (assuming a 76bp read):
30M*3000N*46M. In the genome, this alignment “spans” 3076 bp, yet the nucleotides in the sequencing
read only align “cover” 76bp. Without the -split option, coverage or overlaps would be reported for the
entire 3076bp span of the alignment. However, with the -split option, coverage or overlaps will only
be reported for the portions of the read that overlap the exons (i.e. 30bp on one exon, and
46bp on the other).
Using the -split option with bamToBed causes “spliced/split” alignments to be reported in BED12
format. Using the -split option with bed12tobed6 causes “blocked” BED12 features to be reported in
BED6 format.
Writing uncompressed BAM output.¶
When working with a large BAM file using a complex set of tools in a pipe/stream, it is advantageous
to pass uncompressed BAM output to each downstream program. This minimizes the amount of time
spent compressing and decompressing output from one program to the next. All bedtools that create
BAM output (e.g. intersect, window) will now optionally create uncompressed BAM output
using the -ubam option.
Implementation and algorithmic notes.¶
bedtools was implemented in C++ and makes extensive use of data structures and fundamental algorithms from the Standard Template Library (STL). Many of the core algorithms are based upon the genome binning algorithm described in the original UCSC Genome Browser paper (Kent et al, 2002). The tools have been designed to inherit core data structures from central source files, thus allowing rapid tool development and deployment of improvements and corrections. Support for BAM files is made possible through Derek Barnett’s elegant C++ API called BamTools.
Installation¶
bedtools is intended to run in a “command line” environment on UNIX, LINUX
and Apple OS X operating systems. Installing bedtools involves either
downloading the source code and compiling it manually, or installing stable
release from package managers such as
homebrew (for OS X).
Installing stable releases¶
Downloading a pre-compiled binary¶
Note
- The following approach will only work for Linux (non-OSX) systems.
Starting with release 2.28.0, we provide statically-linked binaries thast should work right away on Linux systems. Go to the releases page and look for the static binary named bedtools.static.binary. Right click on it, get the URL, then download it with wget or curl and you should be good to go.
As an example. First, download bedtools.static.binary from the latest release here
mv bedtools.static.binary bedtools
chmod a+x bedtools
Compiling from source via Github¶
Stable, versioned releases of bedtools are made available on Github at the
bedtools2 repository under
the releases tab.
The following commands will install bedtools in a local directory on an UNIX or OS X machine.
Note
1. The bedtools Makefiles utilize the GCC compiler. One should edit the Makefiles accordingly if one wants to use a different compiler.
$ wget https://github.com/arq5x/bedtools2/releases/download/v2.29.1/bedtools-2.29.1.tar.gz
$ tar -zxvf bedtools-2.29.1.tar.gz
$ cd bedtools2
$ make
At this point, one should copy the binaries in ./bin/ to either
usr/local/bin/ or some other repository for commonly used UNIX tools in
your environment. You will typically require administrator (e.g. “root” or
“sudo”) privileges to copy to usr/local/bin/. If in doubt, contact you
system administrator for help.
Installing with package managers¶
In addition, stable releases of bedtools are also available through package
managers such as homebrew (for OS X),
apt-get and yum.
Fedora/Centos. Adam Huffman has created a Red Hat package for bedtools so that one can easily install the latest release using “yum”, the Fedora package manager. It should work with Fedora 13, 14 and EPEL5/6 ( for Centos, Scientific Linux, etc.).
yum install BEDTools
Debian/Ubuntu. Charles Plessy also maintains a Debian package for bedtools that is likely to be found in its derivatives like Ubuntu. Many thanks to Charles for doing this.
apt-get install bedtools
Homebrew. Carlos Borroto has made BEDTools available on the bedtools package manager for OSX.
brew tap homebrew/science
brew install bedtools
MacPorts. Alternatively, the MacPorts ports system can be used to install BEDTools on OSX.
port install bedtools
Development versions¶
The development version of bedtools is maintained in a Github
repository. Bug fixes are addressed
in this repository prior to release, so there may be situations where you will
want to use a development version of bedtools prior to its being promoted to
a stable release. One would either clone the repository with git, as
follows and then compile the source code as describe above:
git clone https://github.com/arq5x/bedtools2.git
or, one can download the source code as a .zip file using the Github
website. Once the zip file is downloaded and uncompressed with the unzip
command, one can compile and install using the instructions above.
Quick start¶
Install bedtools¶
curl http://bedtools.googlecode.com/files/BEDTools.<version>.tar.gz > BEDTools.tar.gz
tar -zxvf BEDTools.tar.gz
cd BEDTools
make
sudo cp bin/* /usr/local/bin/
Use bedtools¶
Below are examples of typical bedtools usage. Using the “-h” option with any bedtools will report a list of all command line options.
Report the base-pair overlap between the features in two BED files.
bedtools intersect -a reads.bed -b genes.bed
Report those entries in A that overlap NO entries in B. Like “grep -v”
bedtools intersect -a reads.bed -b genes.bed -v
Read BED A from STDIN. Useful for stringing together commands. For example, find genes that overlap LINEs but not SINEs.
bedtools intersect -a genes.bed -b LINES.bed | \
bedtools intersect -a stdin -b SINEs.bed -v
Find the closest ALU to each gene.
bedtools closest -a genes.bed -b ALUs.bed
Merge overlapping repetitive elements into a single entry, returning the number of entries merged.
bedtools merge -i repeatMasker.bed -n
Merge nearby repetitive elements into a single entry, so long as they are within 1000 bp of one another.
bedtools merge -i repeatMasker.bed -d 1000
General usage¶
Supported file formats¶
BED format¶
As described on the UCSC Genome Browser website (see link below), the browser extensible data (BED) format is a concise and flexible way to represent genomic features and annotations. The BED format description supports up to 12 columns, but only the first 3 are required for the UCSC browser, the Galaxy browser and for bedtools. bedtools allows one to use the “BED12” format (that is, all 12 fields listed below). However, only intersectBed, coverageBed, genomeCoverageBed, and bamToBed will obey the BED12 “blocks” when computing overlaps, etc., via the “-split” option. For all other tools, the last six columns are not used for any comparisons by the bedtools. Instead, they will use the entire span (start to end) of the BED12 entry to perform any relevant feature comparisons. The last six columns will be reported in the output of all comparisons.
The file description below is modified from: http://genome.ucsc.edu/FAQ/FAQformat#format1.
- chrom - The name of the chromosome on which the genome feature exists.
- Any string can be used. For example, “chr1”, “III”, “myChrom”, “contig1112.23”.
- This column is required.
- start - The zero-based starting position of the feature in the chromosome.
- The first base in a chromosome is numbered 0.
- The start position in each BED feature is therefore interpreted to be 1 greater than the start position listed in the feature. For example, start=9, end=20 is interpreted to span bases 10 through 20,inclusive.
- This column is required.
- end - The one-based ending position of the feature in the chromosome.
- The end position in each BED feature is one-based. See example above.
- This column is required.
- name - Defines the name of the BED feature.
- Any string can be used. For example, “LINE”, “Exon3”, “HWIEAS_0001:3:1:0:266#0/1”, or “my_Feature”.
- This column is optional.
- score - The UCSC definition requires that a BED score range from 0 to 1000, inclusive. However, bedtools allows any string to be stored in this field in order to allow greater flexibility in annotation features. For example, strings allow scientific notation for p-values, mean enrichment values, etc. It should be noted that this flexibility could prevent such annotations from being correctly displayed on the UCSC browser.
- Any string can be used. For example, 7.31E-05 (p-value), 0.33456 (mean enrichment value), “up”, “down”, etc.
- This column is optional.
- strand - Defines the strand - either ‘+’ or ‘-‘.
- This column is optional.
- thickStart - The starting position at which the feature is drawn thickly.
- Allowed yet ignored by bedtools.
- thickEnd - The ending position at which the feature is drawn thickly.
- Allowed yet ignored by bedtools.
- itemRgb - An RGB value of the form R,G,B (e.g. 255,0,0).
- Allowed yet ignored by bedtools.
- blockCount - The number of blocks (exons) in the BED line.
- Allowed yet ignored by bedtools.
- blockSizes - A comma-separated list of the block sizes.
- blockStarts - A comma-separated list of block starts.
- Allowed yet ignored by bedtools.
bedtools requires that all BED input files (and input received from stdin) are tab-delimited. The following types of BED files are supported by bedtools:
- BED3: A BED file where each feature is described by chrom, start, and end.
For example:chr1 11873 14409
- BED4: A BED file where each feature is described by chrom, start, end, and name.
For example:chr1 11873 14409 uc001aaa.3
- BED5: A BED file where each feature is described by chrom, start, end, name, and score.
For example:chr1 11873 14409 uc001aaa.3 0
- BED6: A BED file where each feature is described by chrom, start, end, name, score, and strand.
For example:chr1 11873 14409 uc001aaa.3 0 +
BED12: A BED file where each feature is described by all twelve columns listed above.
For example:
chr1 11873 14409 uc001aaa.3 0 + 11873 11873 0 3 354,109,1189, 0,739,1347,
BEDPE format¶
We have defined a new file format, the browser extensible data paired-end (BEDPE) format, in order to concisely describe disjoint genome features, such as structural variations or paired-end sequence alignments. We chose to define a new format because the existing “blocked” BED format (a.k.a. BED12) does not allow inter-chromosomal feature definitions. In addition, BED12 only has one strand field, which is insufficient for paired-end sequence alignments, especially when studying structural variation.
The BEDPE format is described below. The description is modified from: http://genome.ucsc.edu/FAQ/FAQformat#format1.
- chrom1 - The name of the chromosome on which the first end of the feature exists.
- Any string can be used. For example, “chr1”, “III”, “myChrom”, “contig1112.23”.
- This column is required.
- Use “.” for unknown.
- start1 - The zero-based starting position of the first end of the feature on chrom1.
- The first base in a chromosome is numbered 0.
- As with BED format, the start position in each BEDPE feature is therefore interpreted to be 1 greater than the start position listed in the feature. This column is required.
- Use -1 for unknown.
- end1 - The one-based ending position of the first end of the feature on chrom1.
- The end position in each BEDPE feature is one-based.
- This column is required.
- Use -1 for unknown.
- chrom2 - The name of the chromosome on which the second end of the feature exists.
- Any string can be used. For example, “chr1”, “III”, “myChrom”, “contig1112.23”.
- This column is required.
- Use “.” for unknown.
- start2 - The zero-based starting position of the second end of the feature on chrom2.
- The first base in a chromosome is numbered 0.
- As with BED format, the start position in each BEDPE feature is therefore interpreted to be 1 greater than the start position listed in the feature. This column is required.
- Use -1 for unknown.
- end2 - The one-based ending position of the second end of the feature on chrom2.
- The end position in each BEDPE feature is one-based.
- This column is required.
- Use -1 for unknown.
- name - Defines the name of the BEDPE feature.
- Any string can be used. For example, “LINE”, “Exon3”, “HWIEAS_0001:3:1:0:266#0/1”, or “my_Feature”.
- This column is optional.
- score - The UCSC definition requires that a BED score range from 0 to 1000, inclusive. However, bedtools allows any string to be stored in this field in order to allow greater flexibility in annotation features. For example, strings allow scientific notation for p-values, mean enrichment values, etc. It should be noted that this flexibility could prevent such annotations from being correctly displayed on the UCSC browser.
- Any string can be used. For example, 7.31E-05 (p-value), 0.33456 (mean enrichment value), “up”, “down”, etc.
- This column is optional.
- strand1 - Defines the strand for the first end of the feature. Either ‘+’ or ‘-‘.
- This column is optional.
- Use “.” for unknown.
- strand2 - Defines the strand for the second end of the feature. Either ‘+’ or ‘-‘.
- This column is optional.
- Use “.” for unknown.
- Any number of additional, user-defined fields - bedtools allows one to add as many additional fields to the normal, 10-column BEDPE format as necessary. These columns are merely “passed through” pairToBed and pairToPair and are not part of any analysis. One would use these additional columns to add extra information (e.g., edit distance for each end of an alignment, or “deletion”, “inversion”, etc.) to each BEDPE feature.
- These additional columns are optional.
Entries from an typical BEDPE file:
chr1 100 200 chr5 5000 5100 bedpe_example1 30 + -
chr9 1000 5000 chr9 3000 3800 bedpe_example2 100 + -
Entries from a BEDPE file with two custom fields added to each record:
chr1 10 20 chr5 50 60 a1 30 + - 0 1
chr9 30 40 chr9 80 90 a2 100 + - 2 1
GFF format¶
The GFF format is described on the Sanger Institute’s website (http://www.sanger.ac.uk/resources/software/gff/spec.html). The GFF description below is modified from the definition at this URL. All nine columns in the GFF format description are required by bedtools.
- seqname - The name of the sequence (e.g. chromosome) on which the feature exists.
- Any string can be used. For example, “chr1”, “III”, “myChrom”, “contig1112.23”.
- This column is required.
- source - The source of this feature. This field will normally be used to indicate the program making the prediction, or if it comes from public database annotation, or is experimentally verified, etc.
- This column is required.
- feature - The feature type name. Equivalent to BED’s name field.
- Any string can be used. For example, “exon”, etc.
- This column is required.
- start - The one-based starting position of feature on seqname.
- This column is required.
- bedtools accounts for the fact the GFF uses a one-based position and BED uses a zero-based start position.
- end - The one-based ending position of feature on seqname.
- This column is required.
- score - A score assigned to the GFF feature. Like BED format, bedtools allows any string to be stored in this field in order to allow greater flexibility in annotation features. We note that this differs from the GFF definition in the interest of flexibility.
- This column is required.
- strand - Defines the strand. Use ‘+’, ‘-’ or ‘.’
- This column is required.
- frame - The frame of the coding sequence. Use ‘0’, ‘1’, ‘2’, or ‘.’.
- This column is required.
- attribute - Taken from http://www.sanger.ac.uk/resources/software/gff/spec.html: From version 2 onwards, the attribute field must have an tag value structure following the syntax used within objects in a .ace file, flattened onto one line by semicolon separators. Free text values must be quoted with double quotes. Note: all non-printing characters in such free text value strings (e.g. newlines, tabs, control characters, etc) must be explicitly represented by their C (UNIX) style backslash-escaped representation (e.g. newlines as ‘n’, tabs as ‘t’). As in ACEDB, multiple values can follow a specific tag. The aim is to establish consistent use of particular tags, corresponding to an underlying implied ACEDB model if you want to think that way (but acedb is not required).
- This column is required.
An entry from an example GFF file :
seq1 BLASTX similarity 101 235 87.1 + 0 Target "HBA_HUMAN" 11 55 ;
E_value 0.0003 dJ102G20 GD_mRNA coding_exon 7105 7201 . - 2 Sequence
"dJ102G20.C1.1"
Genome file format¶
Some of the bedtools (e.g., genomeCoverageBed, complementBed, slopBed) need to know the size of the chromosomes for the organism for which your BED files are based. When using the UCSC Genome Browser, Ensemble, or Galaxy, you typically indicate which which species/genome build you are working. The way you do this for bedtools is to create a “genome” file, which simply lists the names of the chromosomes (or scaffolds, etc.) and their size (in basepairs).
Genome files must be tab-delimited and are structured as follows (this is an example for C. elegans):
chrI 15072421
chrII 15279323
...
chrX 17718854
chrM 13794
bedtools includes pre-defined genome files for human and mouse in the /genomes directory included in the bedtools distribution.
One can also create a suitable genome file by running samtools faidx on the appropriate FASTA reference genome. Then use the resulting .fai file as a genome file, as bedtools will only care about the first two columns, which define the chromosome name and length. For example:
# download GRCh38
wget ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/GRCh38_reference_genome/GRCh38_full_analysis_set_plus_decoy_hla.fa
# create an index of it
samtools faidx GRCh38_full_analysis_set_plus_decoy_hla.fa
# use the .fai index as a genome file with bedtools
bedtools complement my.grch38.bed -g GRCh38_full_analysis_set_plus_decoy_hla.fa.fai
SAM/BAM format¶
The SAM / BAM format is a powerful and widely-used format for storing sequence alignment data (see
http://samtools.sourceforge.net/ for more details). It has quickly become the standard format to which
most DNA sequence alignment programs write their output. Currently, the following bedtools
support input in BAM format: intersect, window, coverage, genomecov,
pairtobed, bamtobed. Support for the BAM format in bedtools allows one to (to name a few):
compare sequence alignments to annotations, refine alignment datasets, screen for potential mutations
and compute aligned sequence coverage.
VCF format¶
The Variant Call Format (VCF) was conceived as part of the 1000 Genomes Project as a standardized means to report genetic variation calls from SNP, INDEL and structural variant detection programs (see http://www.1000genomes.org/wiki/doku.php?id=1000_genomes:analysis:vcf4.0 for details). bedtools now supports the latest version of this format (i.e, Version 4.0). As a result, bedtools can be used to compare genetic variation calls with other genomic features.
Release History¶
Version 2.30.0 (23-Dec-2021)¶
- Thanks to Hao Hou (github: @38), we have substantial improvements in the speed associated with parsing input files and in printing results. It turns our that these tasks consume a large proportion of run time, especially as input files increase in size. These changes result in a 2-3X improvement in speed, depending on input types, options, etc.
- Thanks to John Marshall (github: @jmarshall), who improved the stability and cleanliness of the code used for random number generation. These changes also squash a bug that arises on Debian systems.
- John Marshall cleaned up some lingering data type problems in the slop tool.
- Thanks to @gringer for adding teh -ignoreD option to the genomecov tool, which allows D CIGAR operations to be ignored when calculating coverage. This is useful for long-read technologies with high INDEL error rates.
- Added a fix for a bug that did not properly handle the splitting of intervals in BED12 records with one block.
- Thanks to John Marshall (github: @jmarshall), we have addressed numerical instability issues in the fisher tool.
- Thanks to Hao Hou (github: @38), reference genomes can be read as an environment variable (CRAM_REFERENCE) when using CRAM input files.
- Added a -rna option to the getfasta tool to allow support for RNA genomes.
- Thanks to Hao Hou (github: @38), we fixed input file format detection bugs arising in ZSH.
- Thanks to Josh Shapiro (github:@jashapiro) for clarifying a confusing inconcistency in the documentation for the coverage tool.
- Thanks to Hao Hou (github: @38), we suppressed unnecessary warnings when reading GZIPP’ed files.
- Thanks to Hao Hou (github: @38), we fixed an overflow bug in the shuffle tool.
- Thanks to Hao Hou (github: @38), we fixed an data type bug in the shift tool.
- Thanks to John Marshall (github: @jmarshall) and Hao Hou (github: @38), we have cleaned up the internal support for htslib.
Version 2.29.2 (17-Dec-2019)¶
- Fixed a bug that mistakenly removed a BAM/CRAM header line (sorting criteria).
Version 2.29.1 (9-Dec-2019)¶
- Fixed a bug that now allows blocked intersection to be counted based on unique base pairs of overlap. The resolution for issue 750 in version 2.29.0 mistakenly allowed for fractional overlap to be counted based upon redundant overlap.
- Moved to Github Continuous Integration for automatic testing.
- Fixed a bug that injected erroneous quality values with BAM records had no valid quality values.
- Fixed a bug that destroyed backwards compatibility in the getfasta tool. Thanks to Torsten Seeman for reporting this.
- Fixed a corner case bug in the reldist tool.
- Fixed a bug in the bedtobam tool that caused the last character in read names to be removed.
- Fixed a bug causing a segfault in the jaccard tool.
- Fixed a bug causing a corner case issue in the way coordinates are reported in the flank tool.
Version 2.29.0 (3-Sep-2019)¶
- Added a new -C option to the intersect tool that separately reports the count of intersections observed for each database (-b) file given. Formerly, the -c option reported to sum of all intersections observed across all database files.
- Fixed an important bug in intersect that prevented some split reads from being counted properly with using the -split option with the -f option.
- Fixed a bug in shuffle such that shuffled regions should have the same strand as the chose -incl region.
- Added a new -L option to L`imit the output of the `complement tool to solely the chromosomes that are represented in the -i file.
- Fixed a regression in the multicov tool introduced in 2.28 that caused incorrect counts.
- Added support for multi-mapping reads in the bamtofastq tool.
- Fixed a bug that prevented the “window” tool from properly adding interval “slop” to BAM records.
- Fixed a bug that caused the slop tool to not truncate an interval’s end coordinate when it overlapped the end of a chromosome.
- Added support for the “=” and “X” CIGAR operations to bamtobed.
- Various other minor bug fixes and improvements to documentation.
Version 2.28.0 (23-Mar-2019)¶
- Included support for htslib to enable CRAM support and long-term stability (Thanks to Hao Hou!)
- Included support for genomes with large chromosomes by moving to 64-bit integeres throughout the code base. Thanks to Brent Pedersen and John Marshall!
- We now provide a statically-linked binary for LINUX (not OSX) systems.
- Various minor bug fixes.
Version 2.27.0 (6-Dec-2017)¶
- Fixed a big memory leak and algorithmic flaw in the split option. Thanks to Neil Kindlon!
- Resolved compilation errors on OSX High Sierra. Many thanks to @jonchang!
- Fixed a bug in the shift tool that caused some intervals to exceed the end of the chromosome. Thanks to @wlholtz
- Fixed major bug in groupby that prevented proper functionality.
- Speed improvements to the shuffle tool.
- Bug fixes to the p-value calculation in the fisher tool. Thanks to Brent Pedersen.
- Allow BED headers to start with chrom or chr
- Fixes to the “k-closest” functionality in the closest tool. Thanks to Neil Kindlon.
- Fixes to the output of the freqasc, freqdesc, distinct_sort_num and distinct_sort, and num_desc operations in the groupby tool. Thanks to @ghuls.
- Many minor bug fixes and compilation improvements from Luke Goodsell.
- Added the -fullHeader option to the maskfasta tool. Thanks to @ghuls.
- Many bug fixes and performance improvements from John Marshall.
- Fixed bug in the -N/-f behavior in subtract.
- Full support for .fai files as genome (-g) files.
- Many other minor bug fixes and functionality improvements.
Version 2.26.0 (7-July-2016)¶
- Fixed a major memory leak when using
-sorted. Thanks to Emily Tsang and Stephen Montgomery. - Fixed a bug for BED files containing a single record with no newline. Thanks to @jmarshall.
- Fixed a bug in the contigency table values for thr
fishertool. - The
getfastatool includes name, chromosome and position in fasta headers when the-nameoption is used. Thanks to @rishavray. - Fixed a bug that now forces the
coveragetool to process every record in the-afile. - Fixed a bug preventing proper processing of BED files with consecutive tabs.
- VCF files containing structural variants now infer SV length from either the SVLEN or END INFO fields. Thanks to Zev Kronenberg.
- Resolve off by one bugs when intersecting GFF or VCF files with BED files.
- The
shuffletool now uses roulette wheel sampling to shuffle to-inclregions based upon the size of the interval. Thanks to Zev Kronenberg and Michael Imbeault. - Fixed a bug in
coveragethat prevented correct calculation of depth when using the-splitoption. - The
shuffletool warns when an interval exceeds the maximum chromosome length. - The
complementtool better checks intervals against the chromosome lengths. - Fixes for
stddev,min, andmaxoperations. Thanks to @jmarshall. - Enabled
stdev,sstdev,freqasc, andfreqdescoptions forgroupby. - Allow
-sand-wto be used in any order formakewindows. - Added new
-bedOutoption togetfasta. - The
-roption forces the-Fvalue forintersect. - Add
-pcoption to thegenomecovtool, allowing coverage to be calculated based upon paired-end fragments.
Version 2.25.0 (3-Sept-2015)¶
- Added new -F option that allows one to set the minimum fraction of overlap required for the B interval. This complements the functionality of the -f option.Available for intersect, coverage, map, subtract, and jaccard.
- Added new -e option that allows one to require that the minimum fraction overlap is achieved in either A _OR_ B, not A _AND_ B which is the behavior of the -r option. Available for intersect, coverage, map, subtract, and jaccard.
- Fixed a longstanding bug that prevented genomecov from reporting chromosomes that lack a single interval.
- Modified a src directory called “aux” to “driver” to prevent compilation errors on Windows machines. Thanks very much to John Marshall.
- Fixed a regression that caused the coverage tool to complain if BED files had less than 5 columns.
- Fixed a variable overload bug that prevented compilation on Debian machines.
- Speedups to the groupby tool.
- New -delim option for the groupby tool.
- Fixed a bug in map that prevented strand-specifc overlaps from being reported when using certain BEDPLUS formats.
- Prevented excessive memory usage when not using pre-sorted input.
Version 2.24.0 (27-May-2015)¶
- The coverage tool now takes advantage of pre-sorted intervals via the -sorted option. This allows the coverage tool to be much faster, use far less memory, and report coverage for intervals in their original order in the input file.
- We have changed the behavior of the coverage tool such that it is consistent with the other tools. Specifically, coverage is now computed for the intervals in the A file based on the overlaps with the B file, rather than vice versa.
- The
subtracttool now supports pre-sorted data via the-sortedoption and is therefore much faster and scalable. - The
-nonamecheckoption provides greater tolerance for chromosome labeling when using the-sortedoption. - Support for multiple SVLEN tags in VCF format, and fixed a bug that failed to process SVLEN tags coming at the end of a VCF INFO field.
- Support for reverse complementing IUPAC codes in the
getfastatool. - Provided greater flexibility for “BED+” files, where the first 3 columns are chrom, start, and end, and the remaining columns are free-form.
- We now detect stale FAI files and recreate an index thanks to a fix from @gtamazian.
- New feature from Pierre Lindenbaum allowing the
sorttool to sort files based on the chromosome order in afaidxfile. - Eliminated multiple compilation warnings thanks to John Marshall.
- Fixed bug in handling INS variants in VCF files.
Version 2.23.0 (22-Feb-2015)¶
- Added
-koption to the closest tool to report the k-closest features in one or more -b files. - Added
-fdoption to the closest tool to for the reporting of downstream features in one or more -b files. Requires -D to dictate how “downstream” should be defined. - Added
-fuoption to the closest tool to for the reporting of downstream features in one or more -b files. Requires -D to dictate how “downstream” should be defined. - Pierre Lindenbaum added a new split tool that will split an input file into multiple sub files. Unlike UNIX split, it can balance the chunking of the sub files not just by number of lines, but also by total number of base pairs in each sub file.
- Added a new spacing tool that reports the distances between features in a file.
- Jay Hesselberth added a
-reverseoption to the makewindows tool that reverses the order of the assigned window numbers. - Fixed a bug that caused incorrect reporting of overlap for zero-length BED records. Thanks to @roryk.
- Fixed a bug that caused the map tool to not allow
-bto be specified before-a. Thanks to @semenko. - Fixed a bug in
makewindowsthat mistakenly required-swith-n.
Version 2.22.1 (01-Jan-2015)¶
- When using -sorted with intersect, map, and closest, bedtools can now detect and warn you when your input datasets employ different chromosome sorting orders.
- Fixed multiple bugs in the new, faster closest tool. Specifically, the -iu, -id, and -D options were not behaving properly with the new “sweeping” algorithm that was implemented for the 2.22.0 release. Many thanks to Sol Katzman for reporting these issues and for providing a detailed analysis and example files.
- We FINALLY wrote proper documentation for the closest tool (http://bedtools.readthedocs.org/en/latest/content/tools/closest.html)
- Fixed bug in the tag tool when using -intervals, -names, or -scores. Thanks to Yarden Katz for reporting this.
- Fixed issues with chromosome boundaries in the slop tool when using negative distances. Thanks to @acdaugherty!
- Multiple improvements to the fisher tool. Added a -m option to the fisher tool to merge overlapping intervals prior to comparing overlaps between two input files. Thanks to@brentp
- Fixed a bug in makewindows tool requiring the use of -b with -s.
- Fixed a bug in intersect that prevented -split from detecting complete overlaps with -f 1. Thanks to @tleonardi .
- Restored the default decimal precision to the groupby tool.
- Added the -prec option to the merge and map tools to specific the decimal precision of the output.
Version 2.22.0 (12-Nov-2014)¶
- The “closest” tool now requires sorted files, but this requirement now enables it to simultaneously find the closest intervals from many (not just one) files.
- We now have proper support for “imprecise” SVs in VCF format. This addresses a long standing (sorry) limitation in the way bedtools handles VCF files.
Version 2.21.0 (18-Sep-2014)¶
- Added ability to intersect against multiple -b files in the intersect tool.
- Fixed a bug causing slowdowns in the -sorted option when using -split with very large split alignments.
- Added a new fisher tool to report a P-value associated with the significance of the overlaps between two interval sets. Thanks to @brentp!
- Added a “genome” file for GRCh38. Thanks @martijnvermaat!
- Fixed a bug in the -pct option of the slop tool. Thanks to @brentp!
- Tweak to the Makefile to accomodate Intel compilers. Thanks to @jmarshall.
- Many updates to the docs from the community. Thank you!
Version 2.20.1 (23-May-2014)¶
- Fixed a float rounding bug causing occassional off-by-one issues in the slop added by the
sloptool. Thanks to @slw287r. - Fixed a bug injected in 2.19 arising when files have a single line not ending in a newline. Thanks to @cwarden45.
Version 2.20.0 (22-May-2014)¶
- The
mergetool now supports BAM input.
- The
-n,-nms, and-scoresoptions are deprecated in favor of the new, substantially more flexible,-cand-ooptions. See the docs.- It now supports the
-headeroption.- It now supports the
-Soption.
- The
maptool now supports BAM input. - The
jaccardtool is now ~3 times faster.
- It now supports the
-splitoption.- It now supports the
-soption.- It now supports the
-Soption.
- We have fixed several CLANG compiler issues/ Thanks to John Marshall for the thorough report.
- We added support for “X” and “=” CIGAR operators. Thanks to Pierre Lindenbaum.
- Fixed bugs for empty files.
- Improved the
-incloption in theshuffletool such that the distibution is much more random. - Fixed a bug in
slopwhen very large slop values are used.
Version 2.19.1 (6-Mar-2014)¶
- Bug fix to intersect causing BAM footers to be erroneously written when -b is BAM
- Speedup for the map tool. - http://bedtools.readthedocs.org/en/latest/_images/map-speed-comparo.png
- Map tool now allows multiple columns and operations in a single run. - http://bedtools.readthedocs.org/en/latest/content/tools/map.html#multiple-operations-and-columns-at-the-same-time
Version 2.19.0 (8-Feb-2014)¶
Bug Fixes¶
- Fixed a long standing bug in which the number of base pairs of overlap was incorrectly calculated when using the -wo option with the -split option. Thanks to many for reporting this.
- Fixed a bug in which certain flavors of unmapped BAM alignments were incorrectly rejected in the latest 2.18.* series. Thanks very much to Gabriel Pratt.
Enhancements¶
- Substantially reduced memory usage, especially when dealing with unsorted data. Memory usage ballooned in the 2.18.* series owing to default buffer sizes we were using in a custom string class. We have adjusted this and the memory usage has returned to 2.17.* levels while maintaining speed increases. Thanks so much to Ian Sudberry rightfully complaining about this!
New features¶
- The latest version of the “map” function is ~3X faster than the one available in version 2.17 and 2.18
- The map function now supports the “-split” option, as well as “absmin” and “absmax” operations.
- In addition, it supports multiple chromosome sorting criterion by supplying a genome file that defines the expected chromosome order. Here is an example of how to run map with datasets having chromosomes sorted in “version” order, as opposed to the lexicographical chrom order that is the norm.
Version 2.18.2 (8-Jan-2014)¶
bedtools¶
The changes to bedtools reflect fixes to compilation errors, performance enhancements for smaller files, and a bug fix for BAM files that lack a formal header. Our current focus for the 2.19.* release is is on addressing some standing bug/enhancements and also in updating some of the other more widely used tools (e.g., coverage, map, and substract) to use the new API. We will also continue to look into ways to improve performance while hopefully reducing memory usage for algorithms that work with unsorted data (thanks to Ian Sudberry for the ping!).
pybedtools¶
Ryan Dale has updated pybedtools to accomodate bedtools 2.18.*, added unit tests, and provided new functionality and bug fixes. The details for this release are here: http://pythonhosted.org/pybedtools/changes.html
Version 2.18.1 (16-Dec-2013)¶
Fixes that address compilation errors with CLANG and force compilation of custom BamTools library.
Version 2.18.0 (13-Dec-2013)¶
The Google Code site is deprecated¶
It looks like the Google Code service is going the way of the venerable Google Reader. As such, we are moving the repository and all formal release tarballs to Github. We have started a new repository prosaically named “bedtools2”. The original bedtools repository will remain for historical purposes, but we created a new repository to distinguish the two code bases as they will become rather different over time.
[https://github.com/arq5x/bedtools2](https://github.com/arq5x/bedtools2)
We gutted the core API and algorithms¶
Much of Neil’s hard work has been devoted to completely rewriting the core file/stream writing API to be much more flexible in the adoption of new formats. In addition, he has substantially improved many of the core algorithms for detecting interval intersections.
Improved performance¶
The 2.18.0 release leverages these improvements in the “intersect” tool. Forthcoming releases will see the new API applied to other tools, but we started with intersect as it is the most widely used tool in the suite.
Performance with sorted datasets. The “chromsweep” algorithm we use for detecting intersections is now 60 times faster than when it was first release in version 2.16.2, and is 15 times than the 2.17 release. This makes the algorithm slightly faster that the algorithm used in the bedops bedmap tool. As an example, the following [figure](https://dl.dropboxusercontent.com/u/515640/bedtools-intersect-sorteddata.png) demonstrates the speed when intersecting GENCODE exons against 1, 10, and 100 million BAM alignments from an exome capture experiment. Whereas in version 2.16.2 this wuld have taken 80 minutes, it now takes 80 seconds.
Greater flexibility. In addition, BAM, BED, GFF/GTF, or VCF files are now automatically detected whether they are a file, stream, or FIFO in either compressed or uncompressed form. As such, one now longer has specify -abam when using BAM input as the “A” file with intersect. Moreover, any file type can be used for either the A or
the B file.
Better support for different chromosome sorting criteria¶
Genomic analysis is plagued by different chromosome naming and sorting conventions. Prior to this release,
the -sorted option in the intersect tool required that the chromosomes were sorted in alphanumeric
order (e.g. chr1, chr10, etc. or 1, 10, etc.). Starting with this release, we now simply require by default
that the records are GROUPED by chromosome and that within each chromosome group, the records are sorted by
chromosome position. This will allow greater flexibility.
One problem that can arise however, is if two different files are each grouped by chromosome, yet the two files follow a different chromosome order. In order to detect and enforce the same order, one can explicitly state the expected chromosome order through the use of a genome (aka chromsizes) file. Please see the documentation [here](http://bedtools.readthedocs.org/en/latest/content/tools/intersect.html#sorted-invoke-a-memory-efficient-algorithm-for-very-large-files) and [here](http://bedtools.readthedocs.org/en/latest/content/tools/intersect.html#g-define-an-alternate-chromosome-sort-order-via-a-genome-file) for examples.
New tools¶
1. The jaccard tool. While not exactly new, there have been improvements to the tool and there is finally
documentation. Read more here: http://bedtools.readthedocs.org/en/latest/content/tools/jaccard.html
- The
reldisttool. Details here: http://bedtools.readthedocs.org/en/latest/content/tools/reldist.html
3. The sample tool. Uses reservoir sampling to randomly sample a specified number of records from BAM, BED,
VCF, and GFF/GTF files.
Enhancements¶
- Improvements in the consistency of the output of the
mergetool. Thanks to @kcha.
2. A new -allowBeyondChromEnd option in the shuffle tool. Thanks to @stephenturner.
[docs](http://bedtools.readthedocs.org/en/latest/content/tools/shuffle.html#allowbeyondchromend-allow-records-to-extend-beyond-the-chrom-length)
- A new
-noOverlappingoption that prevents shuffled intervals from overlapping one another. Thanks to @brentp. [docs](http://bedtools.readthedocs.org/en/latest/content/tools/shuffle.html#nooverlapping-prevent-shuffled-intervals-from-overlapping) - Allow the user to specify the maximum number of shuffling attempts via the
-maxTriesoption in theshuffletool. - Various improvements to the documentation provided by manu different users. Thanks to all.
- Added the number of intersections (
n_intersections) to the Jaccard output. Thanks to @brentp. - Various improvements to the
tagtool. - Added the
-N(remove any) option to thesubtracttool.
Version 2.17.0 (3-Nov-2012)¶
New tools¶
We have added a new tool (bedtools “jaccard”) for measuring the Jaccard statistic between two interval files. The Jaccard stat measures the ratio of the length of the intersection over the length of the union of the two sets. In this case, the union is measured as the sum of the lengths of the intervals in each set minus the length of the intersecting intervals. As such, the Jaccard statistic provides a “distance” measure between 0 (no intersections) and 1 (self intersection). The higher the score, the more the two sets of intervals overlap one another. This tool was motivated by Favorov et al, 2012. For more details, see see PMID: 22693437.
We anticipate releasing other statistical measures in forthcoming releases.
New Features & enhancements¶
- The genome file drives the BAM header in “bedtools bedtobam”
- Substantially improvement the performance of the -sorted option in “bedtools intersect” and “bedtools map”. For many applications, bedtools is now nearly as fast as the BEDOPS suite when intersecting pre-sorted data. This improvement is thanks to Neil Kindlon, a staff scientist in the Quinlan lab.
- Tightened the logic for handling split (blocked) BAM and BED records
- Added ranged column selection to “bedtools groupby”. Thanks to Brent Pedersen”
- e.g., formerly “bedtools groupby -g 1,2,3,4,5”; now “-g 1-5”
- “bedtools getfasta” now properly extracts sequences based on blocked (BED12) records (e.g., exons from genes in BED12 format).
- “bedtools groupby” now allows a header line in the input.
- With -N, the user can now force the closest interval to have a different name field in “bedtools closest”
- With -A, the user can now force the subtraction of entire interval when any overlap exists in “bedtools subtract”.
- “bedtools shuffle” can now shuffle BEDPE records.
- Improved random number generation.
- Added -split, -s, -S, -f, -r options to “bedtools multicov”
- Improvements to the regression testing framework.
- Standardized the tag reporting logic in “bedtools bamtobed”
- Improved the auto-detection of VCF format. Thanks to Michael James Clark.
Bug fixes¶
- Fixed a bug in bedtobam’s -bed12 mode.
- Properly include unaligned BAM alignments with “bedtools intersect“‘s -v option.
- Fixed off by one error in “bedtools closest“‘s -d option
4.”bedtools bamtobed” fails properly for non-existent file.
- Corrected missing tab in “bedtools annotate“‘s header.
- Allow int or uint tags in “bedtools bamtobed”
- “bedtools flank” no longer attempts to take flanks prior to the start of a chromosome.
- Eliminated an extraneous tab from “bedtools window” -c.
- Fixed a corner case in the -sorted algorithm.
10.Prevent numeric overflow in “bedtools coverage -hist”
Version 2.14.1-3 (2-Nov-2011)¶
Bug Fixes¶
- Corrected the help for closestBed. It now correctly reads -io instead of -no.
- Fixed regression in closestBed injected in version 2.13.4 whereby B features to the right of an A feature were missed.
New tool¶
- Added the multiIntersectBed tool for reporting common intervals among multiple sorted BED/GFF/VCF files.
Version 2.13.4 (26-Oct-2011)¶
Bug Fixes¶
- The -sorted option (chromsweep) in intersectBed now obeys -s and -S. I had neglected to implement that. Thanks to Paul Ryvkin for pointing this out.
- The -split option was mistakenly splitting of D CIGAR ops.
- The Makefile was not including zlib properly for newer versions of GCC. Thanks to Istvan Albert for pointing this out and providing the solution.
Improvements¶
- Thanks to Jacob Biesinger for a new option (-D) in closestBed that will report _signed_ distances. Moreover, the new option allows fine control over whether the distances are reported based on the reference genome or based on the strand of the A or B feature. Many thanks to Jacob.
- Thanks to some nice analysis from Paul Ryvkin, I realized that the -sorted option was using way too much memory in certain cases where there is a chromosome change in a sorted BED file. This has been corrected.
Version 2.13.3 (30-Sept-2011)¶
Bug Fixes¶
- intersectBed detected, but did not report overlaps when using BAM input and -bed.
Other¶
- Warning that -sorted trusts, but does not enforce that data is actually sorted.
Version 2.13.2 (23-Sept-2011)¶
New algorithm¶
- Preliminary release of the chrom_sweep algorithm.
New options¶
- genomeCoverageBed no longer requires a genome file when working with BAM input. It instead uses the BAM header.
- tagBam now has a -score option for annotating alignments with the BED “scores” field in annotation files. This overrides the default behavior, which is to use the -labels associated with the annotation files passed in on the command line.
Bug fixes¶
- Correct a bug that prevented proper BAM support in intersectBed.
- Improved detection of GFF features with negative coordinates.
Version 2.13.1 (6-Sept-2011)¶
New options¶
- tagBam now has -s and -S options for only annotating alignments with features on the same and opposite strand, respectively.
- tagBam now has a -names option for annotating alignments with the “name” field in annotation files. This overrides the default behavior, which is to use the -labels associated with the annotation files passed in on the command line. Currently, this works well with BED files, but given the limited metadata support for GFF files, annotating with -names and GFF files may not work as well as wished, depending on the type of GFF file used.
Version 2.13.0 (1-Sept-2011)¶
New tools¶
1. tagBam. This tool annotates a BAM file with custom tag fields based on overlaps with BED/GFF/VCF files. For example:
$ tagBam -i aln.bam -files exons.bed introns.bed cpg.bed utrs.bed \
-tags exonic intonic cpg utr \
> aln.tagged.bam
For alignments that have overlaps, you should see new BAM tags like “YB:Z:exonic”, “YB:Z:cpg;utr” 2. multiBamCov. The new tool counts sequence coverage for multiple bams at specific loci defined in a BED/GFF/VCF file. For example:
$ multiBamCov -bams aln.1.bam aln.2.bam aln3.bam -bed exons.bed chr1 861306 861409 SAMD11 1 + 181 280 236 chr1 865533 865718 SAMD11 2 + 249 365 374 chr1 866393 866496 SAMD11 3 + 162 298 322
where the last 3 columns represent the number of alignments overlapping each interval from the three BAM file.
- The following options are available to control which types of alignments are are counted.
-q Minimum mapping quality allowed. Default is 0. -D Include duplicate-marked reads. Default is to count non-duplicates only -F Include failed-QC reads. Default is to count pass-QC reads only -p Only count proper pairs. Default is to count all alignments with MAPQ greater than the -q argument, regardless of the BAM FLAG field.
- nucBed. This new tool profiles the nucleotide content of intervals in a fasta file. The following information will be reported after each original BED/GFF/VCF entry:
- %AT content
- %GC content
- Number of As observed
- Number of Cs observed
- Number of Gs observed
- Number of Ts observed
- Number of Ns observed
- Number of other bases observed
- The length of the explored sequence/interval.
- The sequence extracted from the FASTA file. (optional, if -seq is used)
- The number of times a user defined pattern was observed. (optional, if -pattern is used.)
- For example:
- $ nucBed -fi ~/data/genomes/hg18/hg18.fa -bed simrep.bed | head -3 #1_usercol 2_usercol 3_usercol 4_usercol 5_usercol 6_usercol 7_pct_at 8_pct_gc 9_num_A 10_num_C 11_num_G 12_num_T 13_num_N 14_num_oth 15_seq_len chr1 10000 10468 trf 789 + 0.540598 0.459402 155 96 119 98 0 0 468 chr1 10627 10800 trf 346 + 0.445087 0.554913 54 55 41 23 0 0 173
- One can also report the sequence itself:
- $ nucBed -fi ~/data/genomes/hg18/hg18.fa -bed simrep.bed -seq | head -3 #1_usercol 2_usercol 3_usercol 4_usercol 5_usercol 6_usercol 7_pct_at 8_pct_gc 9_num_A 10_num_C 11_num_G 12_num_T 13_num_N 14_num_oth 15_seq_len 16_seq chr1 10000 10468 trf 789 + 0.540598 0.459402 155 96 119 98 0 0 468 ccagggg… chr1 10627 10800 trf 346 + 0.445087 0.554913 54 55 41 23 0 0 173 TCTTTCA…
- Or, one can count the number of times that a specific pattern occur in the intervals (reported as the last column):
- $ nucBed -fi ~/data/genomes/hg18/hg18.fa -bed simrep.bed -pattern CGTT | head #1_usercol 2_usercol 3_usercol 4_usercol 5_usercol 6_usercol 7_pct_at 8_pct_gc 9_num_A 10_num_C 11_num_G 12_num_T 13_num_N 14_num_oth 15_seq_len 16_user_patt_count chr1 10000 10468 trf 789 + 0.540598 0.459402 155 96 119 98 0 0 468 0 chr1 10627 10800 trf 346 + 0.445087 0.554913 54 55 41 23 0 0 173 0 chr1 10757 10997 trf 434 + 0.370833 0.629167 49 70 81 40 0 0 240 0 chr1 11225 11447 trf 273 + 0.463964 0.536036 44 86 33 59 0 0 222 0 chr1 11271 11448 trf 187 + 0.463277 0.536723 37 69 26 45 0 0 177 0 chr1 11283 11448 trf 199 + 0.466667 0.533333 37 64 24 40 0 0 165 0 chr1 19305 19443 trf 242 + 0.282609 0.717391 17 57 42 22 0 0 138 1 chr1 20828 20863 trf 70 + 0.428571 0.571429 10 7 13 5 0 0 35 0 chr1 30862 30959 trf 79 + 0.556701 0.443299 35 22 21 19 0 0 97 0
New options¶
Support for named pipes and FIFOs.
“-” is now allowable to indicate that data is being sent via stdin.
Multiple tools. Added new -S option to annotateBed, closestBed, coverageBed, intersectBed, pairToBed, subtractBed, and windowBed (-Sm). This new option does the opposite of the -s option: that is, overlaps are only processed if they are on _opposite_ strands. Thanks to Sol Katzman for the great suggestion. Very useful for certain RNA-seq analyses.
coverageBed. Added a new -counts option to coverageBed that only reports the count of overlaps, instead of also computing fractions, etc. This is much faster and uses much less memory.
fastaFromBed. Added a new -full option that uses the full BED entry when naming each output sequence. Also removed the -fo option such that all output is now written to stdout.
- genomeCoverageBed.
- Added new -scale option that allows the coverage values to be scaled by a constant. Useful for normalizing coverage with RPM, RPKM, etc. Thanks to Ryan Dale for the useful suggestion.
- Added new -5, -3, -trackline, -trackopts, and -dz options. Many thanks to Assaf Gordon for these improvements.
- -5: Calculate coverage of 5” positions (instead of entire interval) -3: Calculate coverage of 3” positions (instead of entire interval). -trackline: Adds a UCSC/Genome-Browser track line definition in the first line of the output. -trackopts: rites additional track line definition parameters in the first line. -dz: Report the depth at each genome position with zero-based coordinates, instead of zero-based.
- closestBed. See below, thanks to Brent Pedersen, Assaf Gordon, Ryan Layer and Dan Webster for the helpful discussions.
- closestBed now reports _all_ features in B that overlap A by default. This allows folks to decide which is the “best” overlapping feature on their own. closestBed now has a “-io” option that ignores overlapping features. In other words, it will only report the closest, non-overlapping feature.
An example:
$ cat a.bed chr1 10 20
$ cat b.bed chr1 15 16 chr1 16 40 chr1 100 1000 chr1 200 1000
$ bin/closestBed -a a.bed -b b.bed chr1 10 20 chr1 15 16 chr1 10 20 chr1 16 40
$ bin/closestBed -a a.bed -b b.bed -io chr1 10 20 chr1 100 1000
Updates¶
- Updated to the latest version of BamTools. This allows greater functionality and will facilitate new options and tools in the future.
Bug Fixes¶
- GFF files cannot have zero-length features.
- Corrected an erroneous check on the start coordinates in VCF files. Thanks to Jan Vogel for the correction.
- mergeBed now always reports output in BED format.
- Updated the text file Tokenizer function to yield 15% speed improvement.
- Various tweaks and improvements.
Version 2.12.0 (April-3-2011)¶
New Tool¶
- Added new tool called “flankBed”, which allows one to extract solely the flanking regions that are upstream and downstream of a given feature. Unlike slopBed, flankBed does not include the original feature itself. A new feature is created for each flabking region. For example, imagine the following feature:
chr1 100 200
The following would create features for solely the 10 bp regions flanking this feature. $ bin/flankBed -i a.bed -b 10 -g genomes/human.hg18.genome chr1 90 100 chr1 200 210
In contrast, slopBed would return: bin/slopBed -i a.bed -b 10 -g genomes/human.hg18.genome chr1 90 210
FlankBed has all of the same features as slopBed.
New Features¶
1. Added new “-scores” feature to mergeBed. This allows one to take the sum, min, max, mean, median, mode, or antimode of merged feature scores. In addition, one can use the “collapse” operation to get a comma-separated list of the merged scores. 2. mergeBed now tolerates multiple features in a merged block to have the same feature name. 3. Thanks to Erik Garrison’s “fastahack” library, fastaFromBed now reports its output in the order of the input file. 4. Added a “-n” option to bed12ToBed6, which forces the score field to be the 1-based block number from the original BED12 feature. This is useful for tracking exon numbers, for example. 5. Thanks to Can Alkan, added a new “-mc” option to maskFastaFromBed that allows one to define a custom mask character, such as “X” (-n X).
Bug Fixes¶
- Thanks to Davide Cittaro, intersectBed and windowBed now properly capture unmapped BAM alignments when using the “-v” option.
- ClosestBed now properly handles cases where b.end == a.start
- Thanks to John Marshall, the default constructors are much safer and less buggy.
- Fixed bug in shuffleBed that complained about a lack of -incl and -excl.
- Fixed bug in shuffleBed for features that would go beyond the end of a chromosome.
- Tweaked bedToIgv to make it more Windows friendly.
Version 2.11.2 (January-31-2010)¶
Fixed a coordinate reporting bug in coverageBed. Added “max distance (-d)” argument back to the new implementation of mergeBed.
Version 2.11.0 (January-21-2010)¶
Enhancements:¶
- Support for zero length features (i.e., start = end) - For example, this allows overlaps to be detected with insertions in the reference genome, as reported by dbSNP.
- Both 8 and 9 column GFF files are now supported.
- slopBed can now extend the size of features by a percentage of it’s size (-pct) instead of just a fixed number of bases.
- Two improvements to shuffleBed: 3a. A -f (overlapFraction) parameter that defines the maximum overlap that a randomized feature can have with an -excl feature. That is, if a chosen locus has more than -f overlap with an -excl feature, a new locus is sought. 3b. A new -incl option (thanks to Michael Hoffman and Davide Cittaro) that, defines intervals in which the randomized features should be placed. This is used instead of placing the features randomly in the genome. Note that a genome file is still required so that a randomized feature does not go beyond the end of a chromosome.
- bamToBed can now optionally report the CIGAR string as an additional field.
- pairToPair can now report the entire paired feature from the B file when overlaps are found.
- complementBed now reports all chromosomes, not just those with features in the BED file.
- Improved randomization seeding in shuffleBed. This prevents identical output for runs of shuffleBed that occur in the same second (often the case).
Bug Fixes:¶
- Fixed the “BamAlignmentSupportData is private” compilation issue.
- Fixed a bug in windowBed that caused positions to run off the end of a chromosome.
Major Changes:¶
- The groupBy command is now part of the filo package (https://github.com/arq5x/filo) and will no longer be distributed with BEDTools.
Version 2.10.0 (September-21-2010)¶
New tools¶
1. annotateBed. Annotates one BED/VCF/GFF file with the coverage and number of overlaps observed from multiple other BED/VCF/GFF files. In this way, it allows one to ask to what degree one feature coincides with multiple other feature types with a single command. For example, the following will annotate the fraction of the variants in variants.bed that are covered by genes, conservaed regions and know variation, respectively. $ annotateBed -i variants.bed -files genes.bed conserv.bed known_var.bed
This tool was suggested by Can Alkan and was motivated by the example source code that he kindly provided.
New features¶
- New frequency operations (freqasc and freqdesc) added to groupBy. These operations report a histogram of the frequency that each value is observed in a given column.
- Support for writing uncompressed bam with the -ubam option.
- Shorthand arguments for groupBy (-g eq. -grp, -c eq. -opCols, -o eq. -opCols).
- In addition, all BEDTools that require only one main input file (the -i file) will assume that input is coming from standard input if the -i parameter is ignored.
Bug fixes¶
- Increased the precision of the output from groupBy.
Version 2.9.0 (August-16-2010)¶
New tools¶
- unionBedGraphs. This is a very powerful new tool contributed by Assaf Gordon from CSHL. It will combine/merge multiple BEDGRAPH files into a single file, thus allowing comparisons of coverage (or any text-value) across multiple samples.
New features¶
- New “distance feature” (-d) added to closestBed by Erik Arner. In addition to finding the closest feature to each feature in A, the -d option will report the distance to the closest feature in B. Overlapping features have a distance of 0.
- New “per base depth feature” (-d) added to coverageBed. This reports the per base coverage (1-based) of each feature in file B based on the coverage of features found in file A. For example, this could report the per-base depth of sequencing reads (-a) across each capture target (-b).
Bug Fixes¶
- Fixed bug in closestBed preventing closest features from being found for A features with start coordinates < 2048000. Thanks to Erik Arner for pointing this out.
- Fixed minor reporting annoyances in closestBed. Thanks to Erik Arner.
- Fixed typo/bug in genomeCoverageBed that reported negative coverage owing to numeric overflow. Thanks to Alexander Dobin for the detailed bug report.
- Fixed other minor parsing and reporting bugs/annoyances.
Version 2.8.3 (July-25-2010)¶
- Fixed bug that caused some GFF files to be misinterpreted as VCF. This prevented the detection of overlaps.
- Added a new “-tag” option in bamToBed that allows one to choose the _numeric_ tag that will be used to populate the score field. For example, one could populate the score field with the alignment score with “-tag AS”.
- Updated the BamTools API.
Version 2.8.2 (July-18-2010)¶
- Fixed a bug in bedFile.h preventing GFF strands from being read properly.
- Fixed a bug in intersectBed that occasionally caused spurious overlaps between BAM alignments and BED features.
- Fixed bug in intersectBed causing -r to not report the same result when files are swapped.
- Added checks to groupBy to prevent the selection of improper opCols and groups.
- Fixed various compilation issues, esp. for groupBy, bedToBam, and bedToIgv.
- Updated the usage statements to reflect bed/gff/vcf support.
- Added new fileType functions for auto-detecting gzipped or regular files. Thanks to Assaf Gordon.
Version 2.8.1 (July-05-2010)¶
- Added bedToIgv.
Version 2.8.0 (July-04-2010)¶
- Proper support for “split” BAM alignments and “blocked” BED (aka BED12) features. By using the “-split” option, intersectBed, coverageBed, genomeCoverageBed, and bamToBed will now correctly compute overlaps/coverage solely for the “split” portions of BAM alignments or the “blocks” of BED12 features such as genes.
- Added native support for the 1000 Genome Variant Calling Format (VCF) version 4.0.
- New bed12ToBed6 tool. This tool will convert each block of a BED12 feature into discrete BED6 features.
- Useful new groupBy tool. This is a very useful new tool that mimics the “groupBy” clause in SQL. Given a file or stream that is sorted by the appropriate “grouping columns”, groupBy will compute summary statistics on another column in the file or stream. This will work with output from all BEDTools as well as any other tab-delimited file or stream. Example summary operations include: sum, mean, stdev, min, max, etc. Please see the help for the tools for examples. The functionality in groupBy was motivated by helpful discussions with Erik Arner at Riken.
- Improvements to genomeCoverageBed. Applied several code improvements provided by Gordon Assaf at CSHL. Most notably, beyond the several efficiency and organizational changes he made, he include a “-strand” option which allows one to specify that coverage should only be computed on either the “+” or the “-” strand.
- Fixed a bug in closestBed found by Erik Arner (Riken) which incorrectly reported “null” overlaps for features that did not have a closest feature in the B file.
- Fixed a careless bug in slopBed also found by Erik Arner (Riken) that caused an infinite loop when the “-excl” option was used.
- Reduced memory consumption by ca. 15% and run time by ca. 10% for most tools.
- Several code-cleanliness updates such as templated functions and common tyedefs.
- Tweaked the genome binning approach such that 16kb bins are the most granular.
Version 2.7.1 (May-06-2010)¶
Fixed a typo that caused some compilers to fail on closestBed.
Version 2.7.0 (May-05-2010)¶
General: 1. “Gzipped” BED and GFF files are now supported as input by all BEDTools. Such files must end in “.gz”. 2. Tools that process BAM alignments now uniformly compute an ungapped alignment end position based on the BAM CIGAR string. Specifically, “M”, “D” and “N” operations are observed when computing the end position. 3. bamToBed requires the BAM file to be sorted/grouped by read id when creating BEDPE output. This allows the alignments end coordinate for each end of the pair to be properly computed based on its CIGAR string. The same requirement applies to pairToBed. 4. Updated manual. 5. Many silent modifications to the code that improve clarity and sanity-checking and facilitate future additions/modifications.
New Tools: 1. bedToBam. This utility will convert BED files to BAM format. Both “blocked” (aka BED12) and “unblocked” (e.g. BED6) formats are acceptable. This allows one to, for example, compress large BED files such as dbSNP into BAM format for efficient visualization.
- Changes to existing tools:
- intersectBed
- Added -wao option to report 0 overlap for features in A that do not intersect any features in B. This is an extension of the -wo option.
- bamToBed
- Requires that BAM input be sorted/grouped by read name.
- pairToBed
- Requires that BAM input be sorted/grouped by read name.
- Allows use of minimum mapping quality or total edit distance for score field.
- windowBed
- Now supports BAM input.
- genomeCoverageBed
- -bga option. Thanks to Gordon Assaf for the suggestion.
- Eliminated potential seg fault.
- Acknowledgements:
- Gordon Assaf: for suggesting the -bga option in genomeCoverageBed and for testing the new bedToBam utility.
- Ivan Gregoretti: for helping to expedite the inclusion of gzip support.
- Can Alkan: for suggesting the addition of the -wao option to intersectBed.
- James Ward: for pointing out that bedToBam did not need to create “dummy” seq and qual entries.
Version 2.6.1 (Mar-29-2010)¶
- Fixed a careless command line parsing bug in coverageBed.
Version 2.6.0 (Mar-23-2010)¶
Specific improvements / additions to tools¶
- intersectBed. Added an option (-wo) that reports the number of overlapping bases for each intersection b/w A and B files. Not sure why this wasn’t added sooner; it’s obvious.
2. coverageBed - native BAM support - can now report a histogram (-hist) of coverage for each feature in B. Useful for exome sequencing projects, for example. Thanks for the excellent suggestion from Jose Bras - faster
3. genomeCoverageBed - native BAM support - can now report coverage in BEDGRAPH format (-bg). Thanks for the code and great suggestion from Gordon Assaf, CSHL.
4. bamToBed - support for “blocked” BED (aka BED12) format. This facilitates the creation of BED entries for “split” alignments (e.g. RNAseq or SV). Thanks to Ann Loraine, UNCC for test data to support this addition.
5. fastaFromBed - added the ability to extract sequences from a FASTA file according to the strand in the BED file. That is, when “-” the extracted sequence is reverse complemented. Thanks to Thomas Doktor, U. of Southern Denmark for the code and suggestion.
6. *NEW* overlap - newly added tool for computing the overlap/distance between features on the same line.For example:
$ cat test.out chr1 10 20 A chr1 15 25 B chr1 10 20 C chr1 25 35 D $ cat test.out | overlaps -i stdin -cols 2,3,6,7 chr1 10 20 A chr1 15 25 B 5 chr1 10 20 C chr1 25 35 D -5
Bug fixes¶
- Fixed a bug in pairToBed when comparing paired-end BAM alignments to BED annotations and using the “notboth” option.
- Fixed an idiotic bug in intersectBed that occasionally caused segfaults when blank lines existed in BED files.
- Fixed a minor bug in mergeBed when using the -nms option.
General changes¶
- Added a proper class for genomeFiles. The code is much cleaner and the tools are less sensitive to minor problems with the formatting of genome files. Per Gordon Assaf’s wise suggestion, the tools now support “chromInfo” files directly downloaded from UCSC. Thanks Gordon—I disagreed at first, but you were right.
- Cleaned up some of the code and made the API a bit more streamlined. Will facilitate future tool development, etc.
Version 2.5.4 (Mar-3-2010)¶
- Fixed an insidious bug that caused malform BAM output from intersectBed and pairToBed. The previous BAM files worked fine with samtools as BAM input, but when piped in as SAM, there was an extra tab that thwarted conversion from SAM back to BAM. Many thanks to Ivan Gregoretti for reporting this bug. I had never used the BAM output in this way and thus never caught the bug!
Version 2.5.3 (Feb-19-2010)¶
- Fixed bug to “re-allow” track and “browser” lines.
- Fixed bug in reporting BEDPE overlaps.
- Fixed bug when using type “notboth” with BAM files in pairToBed.
- When comparing BAM files to BED/GFF annotations with intersectBed or pairToBed, the __aligned__ sequence is used, rather than the __original__ sequence.
- Greatly increased the speed of pairToBed when using BAM alignments.
- Fixed a bug in bamToBed when reporting edit distance from certain aligners.
Version 2.5.2 (Feb-2-2010)¶
- The start and end coordinates for BED and BEDPE entries created by bamToBed are now based on the __aligned__ sequence, rather than the original sequence. It’s obvious, but I missed it originally…sorry.
- Added an error message to mergeBed preventing one from using “-n” and “-nms” together.
- Fixed a bug in pairToBed that caused neither -type “notispan” nor “notospan” to behave as described.
Version 2.5.1 (Jan-28-2010)¶
- Fixed a bug in the new GFF/BED determinator that caused a segfault when start = 0.
Version 2.5.0 (Jan-27-2010)¶
- Added support for custom BED fields after the 6th column.
- Fixed a command line parsing bug in pairToBed.
- Improved sanity checking.
Version 2.4.2 (Jan-23-2010)¶
- Fixed a minor bug in mergeBed when -nms and -s were used together.
- Improved the command line parsing to prevent the occasional segfault.
Version 2.4.1 (Jan-12-2010)¶
- Updated BamTools libraries to remove some compilation issues on some systems/compilers.
Version 2.4.0 (Jan-11-2010)¶
- Added BAM support to intersectBed and pairToBed
- New bamToBed feature.
- Added support for GFF features
- Added support for “blocked” BED format (BED12)
- Wrote complete manual and included it in distribution.
- Fixed several minor bugs.
- Cleaned up code and improved documentation.
Version 2.3.3 (12/17/2009)¶
Rewrote complementBed to use a slower but much simpler approach. This resolves several bugs with the previous logic.
Version 2.3.2 (11/25/2009)¶
- Fixed a bug in subtractBed that prevent a file from subtracting itself when the following is used:
- $ subtractBed -a test.bed -b test.bed
Version 2.3.1 (11/19/2009)¶
Fixed a typo in closestBed that caused all nearby features to be returned instead of just the closest one.
Version 2.3.0 (11/18/2009)¶
- Added four new tools:
- shuffleBed. Randomly permutes the locations of a BED file among a genome. Useful for testing for significant overlap enrichments.
- slopBed. Adds a requested number of base pairs to each end of a BED feature. Constrained by the size of each chromosome.
- maskFastaFromBed. Masks a FASTA file based on BED coordinates. Useful making custom genome files from targeted capture experiment, etc.
- pairToPair. Returns overlaps between two paired-end BED files. This is great for finding structural variants that are private or shared among samples.
- Increased the speed of intersectBed by nearly 50%.
- Improved corrected some of the help messages.
- Improved sanity checking for BED entries.
Version 2.2.4 (10/27/2009)¶
1. Updated the mergeBed documentation to describe the -names option which allows one to report the names of the features that were merged (separated by semicolons).
Version 2.2.3 (10/23/2009)¶
1. Changed windowBed to optionally define “left” and “right” windows based on strand. For example by default, -l 100 and -r 500 will add 100 bases to the left (lower coordinates) of a feature in A when scanning for hits in B and 500 bases to the right (higher coordinates).
However if one chooses the -sw option (windows bases on strandedness), the behavior changes. Assume the above example except that a feature in A is on the negative strand (“-“). In this case, -l 100, -r 500 and -sw will add 100 bases to the right (higher coordinates) and 500 bases to the left (lower coordinates).
In addition, there is a separate option (-sm) that can optionally force hits in B to only be tracked if they are on the same strand as A.
*NOTE: This replaces the previous -s option and may affect existing pipelines*.
Version 2.2.2 (10/20/2009)¶
- Improved the speed of genomeCoverageBed by roughly 100 fold. The memory usage is now less than 2.0 Gb.
Version 2.2.1¶
1. Fixed a very obvious bug in subtractBed that caused improper behavior when a feature in A was overlapped by more than one feature in B. Many thanks to folks in the Hannon lab at CSHL for pointing this out.
Version 2.2.0¶
Notable changes in this release¶
- coverageBed will optionally only count features in BED file A (e.g. sequencing reads) that overlap with
the intervals/windows in BED file B on the same strand. This has been requested several times recently and facilitates CHiP-Seq and RNA-Seq experiments.
- intersectBed can now require a minimum __reciprocal__ overlap between intervals in BED A and BED B. For example,
previously, if one used -f 0.90, it required that a feature in B overlap 90% of the feature in A for the “hit” to be reported. If one adds the -r (reciprocal) option, the hit must also cover 90% of the feature in B. This helps to exclude overlaps between say small features in A and large features in B:
A ========== B ******************************************************
-f 0.50 (Reported), whereas -f 0.50 -r (Not reported)
- The score field has been changed to be a string. While this deviates from the UCSC definition, it allows one to track
much more meaningful information about a feature/interval. For example, score could now be:
7.31E-05 (a p-value) 0.334577 (mean enrichment) 2:2.2:40:2 (several values encoded in a string)
- closestBed now, by default, reports __all__ intervals in B that overlap equally with an interval in A. Previously, it
merely reported the first such feature that appeared in B. Here’s a cartoon explaining the difference.
Several other minor changes to the algorithms have been made to increase speed a bit.
Version 2.1.2¶
- Fixed yet another bug in the parsing of “track” or “browser” lines. Sigh…
- Change the “score” column (i.e. column 5) to b stored as a string. While this deviates from the UCSC convention, it allows significantly more information to be packed into the column.
Version 2.1.1¶
- Added limits.h to bedFile.h to fix compilation issues on some systems.
- Fixed bug in testing for “track” or “browser” lines.
Version 2.1.0¶
- Fixed a bug in peIntersectBed that prevented -a from being correctly handled when passed via stdin.
- Added new functionality to coverageBed that calculates the density of coverage.
- Fixed bug in geneomCoverageBed.
Version 2.0.1¶
- Added the ability to retain UCSC browser track/browser headers in BED files.
Version 2.0¶
- Sped up the file parsing. ~10-20% increase in speed.
- Created reportBed() as a common method in the bedFile class. Cleans up the code quite nicely.
- Added the ability to compare BED files accounting for strandedness.
- Paired-end intersect.
- Fixed bug that prevented overlaps from being reported when the overlap fraction requested is 1.0
Version 1.2, 04/27/2009.¶
- Added subtractBed.
- Fixed bug that prevented “split” overlaps from being reported.
- Prevented A from being reported if >=1 feature in B completely spans it.
- Added linksBed.
- Added the ability to define separate windows for upstream and downstream to windowBed.
Version 1.1, 04/23/2009.¶
Initial release.
The BEDTools suite¶
bedtools consists of a suite of sub-commands that are invoked as follows:
bedtools [sub-command] [options]
For example, to intersect two BED files, one would invoke the following:
bedtools intersect -a a.bed -b b.bed
The full list of bedtools sub-commands.¶
annotate¶
bedtools annotate, well, annotates one BED/VCF/GFF file with the coverage
and number of overlaps observed from multiple other BED/VCF/GFF files.
In this way, it allows one to ask to what degree one feature coincides with
multiple other feature types with a single command.
Usage and option summary¶
Usage:
bedtools annotate [OPTIONS] -i <BED/GFF/VCF> -files FILE1 FILE2 FILE3 ... FILEn
(or):
annotateBed [OPTIONS] -i <BED/GFF/VCF> -files FILE1 FILE2 FILE3 ... FILEn
| Option | Description |
|---|---|
| -names | A list of names (one per file) to describe each file in -i. These names will be printed as a header line. |
| -counts | Report the count of features in each file that overlap -i. Default behavior is to report the fraction of -i covered by each file. |
| -both | Report the count of features followed by the % coverage for each annotation file. Default is to report solely the fraction of -i covered by each file. |
| -s | Force strandedness. That is, only include hits in A that overlap B on the same strand. By default, hits are included without respect to strand. |
| -S | Require different strandedness. That is, only report hits in B that overlap A on the _opposite_ strand. By default, overlaps are reported without respect to strand. |
Default behavior - annotate one file with coverage from others.¶
By default, the fraction of each feature covered by each annotation file is reported after the complete feature in the file to be annotated.
$ cat variants.bed
chr1 100 200 nasty 1 -
chr2 500 1000 ugly 2 +
chr3 1000 5000 big 3 -
$ cat genes.bed
chr1 150 200 geneA 1 +
chr1 175 250 geneB 2 +
chr3 0 10000 geneC 3 -
$ cat conserve.bed
chr1 0 10000 cons1 1 +
chr2 700 10000 cons2 2 -
chr3 4000 10000 cons3 3 +
$ cat known_var.bed
chr1 0 120 known1 -
chr1 150 160 known2 -
chr2 0 10000 known3 +
$ bedtools annotate -i variants.bed -files genes.bed conserve.bed known_var.bed
chr1 100 200 nasty 1 - 0.500000 1.000000 0.300000
chr2 500 1000 ugly 2 + 0.000000 0.600000 1.000000
chr3 1000 5000 big 3 - 1.000000 0.250000 0.000000
-count Report the count of hits from the annotation files¶
$ bedtools annotate -counts -i variants.bed -files genes.bed conserve.bed known_var.bed
chr1 100 200 nasty 1 - 2 1 2
chr2 500 1000 ugly 2 + 0 1 1
chr3 1000 5000 big 3 - 1 1 0
-both Report both the count of hits and the fraction covered from the annotation files¶
$ bedtools annotate -both -i variants.bed -files genes.bed conserve.bed known_var.bed
#chr start end name score +/- cnt1 pct1 cnt2 pct2 cnt3 pct3
chr1 100 200 nasty 1 - 2 0.500000 1 1.000000 2 0.300000
chr2 500 1000 ugly 2 + 0 0.000000 1 0.600000 1 1.000000
chr3 1000 5000 big 3 - 1 1.000000 1 0.250000 0 0.000000
-s Restrict the reporting to overlaps on the same strand.¶
$ bedtools annotate -s -i variants.bed -files genes.bed conserve.bed known_var.bed
chr1 100 200 nasty 1 - 0.000000 0.000000 0.000000
chr2 500 1000 ugly 2 + 0.000000 0.000000 0.000000
chr3 1000 5000 big 3 - 1.000000 0.000000 0.000000
-S Restrict the reporting to overlaps on the opposite strand.¶
$ bedtools annotate -S -i variants.bed -files genes.bed conserve.bed known_var.bed
chr1 100 200 nasty 1 - 0.500000 1.000000 0.300000
chr2 500 1000 ugly 2 + 0.000000 0.600000 1.000000
chr3 1000 5000 big 3 - 0.000000 0.250000 0.000000
bamtobed¶
bedtools bamtobed is a conversion utility that converts sequence alignments
in BAM format into BED, BED12, and/or BEDPE records.
Usage and option summary¶
Usage:
bedtools bamtobed [OPTIONS] -i <BAM>
(or):
bamToBed [OPTIONS] -i <BAM>
| Option | Description |
|---|---|
| -bedpe | Write BAM alignments in BEDPE format. Only one alignment from paired-end reads will be reported. Specifically, if each mate is aligned to the same chromosome, the BAM alignment reported will be the one where the BAM insert size is greater than zero. When the mate alignments are interchromosomal, the lexicographically lower chromosome will be reported first. Lastly, when an end is unmapped, the chromosome and strand will be set to “.” and the start and end coordinates will be set to -1. By default, this is disabled and the output will be reported in BED format. |
| -mate1 | When writing BEDPE (-bedpe) format, always report mate one as the first BEDPE “block”. |
| -bed12 | Write “blocked” BED (a.k.a. BED12) format. This will convert “spliced” BAM alignments (denoted by the “N” CIGAR operation) to BED12. Forces -split. |
| -split | Report each portion of a “split” BAM (i.e., having an “N” CIGAR operation) alignment as a distinct BED intervals. |
| -splitD | Report each portion of a “split” BAM while obeying both “N” CIGAR and “D” operation. Forces -split. |
| -ed | Use the “edit distance” tag (NM) for the BED score field. Default for BED is to use mapping quality. Default for BEDPE is to use the minimum of the two mapping qualities for the pair. When -ed is used with -bedpe, the total edit distance from the two mates is reported. |
| -tag | Use other numeric BAM alignment tag for BED score. Default for BED is to use mapping quality. Disallowed with BEDPE output. |
| -color | An R,G,B string for the color used with BED12 format. Default is (255,0,0). |
| -cigar | Add the CIGAR string to the BED entry as a 7th column. |
Default behavior¶
By default, each alignment in the BAM file is converted to a 6 column BED. The BED “name” field is comprised of the RNAME field in the BAM alignment. If mate information is available, the mate (e.g., “/1” or “/2”) field will be appended to the name.
$ bedtools bamtobed -i reads.bam | head -3
chr7 118970079 118970129 TUPAC_0001:3:1:0:1452#0/1 37 -
chr7 118965072 118965122 TUPAC_0001:3:1:0:1452#0/2 37 +
chr11 46769934 46769984 TUPAC_0001:3:1:0:1472#0/1 37 -
-tag Set the score field based on BAM tags¶
One can override the choice of the BAM MAPQ as the result BED record’s score
field by using the -tag option. In the example below, we use the -tag
option to select the BAM edit distance (the NM tag) as the score
column in the resulting BED records.
$ bedtools bamtobed -i reads.bam -tag NM | head -3
chr7 118970079 118970129 TUPAC_0001:3:1:0:1452#0/1 1 -
chr7 118965072 118965122 TUPAC_0001:3:1:0:1452#0/2 3 +
chr11 46769934 46769984 TUPAC_0001:3:1:0:1472#0/1 1 -
-bedpe Set the score field based on BAM tags¶
The -bedpe option converts BAM alignments to BEDPE format, thus allowing
the two ends of a paired-end alignment to be reported on a single text line.
Specifically, if each mate is aligned to the same chromosome,
the BAM alignment reported will be the one where the BAM insert size is greater
than zero. When the mate alignments are interchromosomal, the lexicographically
lower chromosome will be reported first. Lastly, when an end is unmapped, the
chromosome and strand will be set to “.” and the start and end coordinates will
be set to -1.
Note
When using this option, it is required that the BAM file is sorted/grouped by the read name. This allows bamToBed to extract correct alignment coordinates for each end based on their respective CIGAR strings. It also assumes that the alignments for a given pair come in groups of twos. There is not yet a standard method for reporting multiple alignments using BAM. bamToBed will fail if an aligner does not report alignments in pairs.
$ bedtools bamtobed -i reads.ba -bedpe | head -3
chr7 118965072 118965122 chr7 118970079 118970129 TUPAC_0001:3:1:0:1452#0 37 + -
chr11 46765606 46765656 chr11 46769934 46769984 TUPAC_0001:3:1:0:1472#0 37 + -
chr20 54704674 54704724 chr20 54708987 54709037 TUPAC_0001:3:1:1:1833#0 37 +
One can easily use samtools and bamToBed together as part of a UNIX pipe. In
this example, we will only convert properly-paired (FLAG == 0x2) reads to
BED format.
$ samtools view -bf 0x2 reads.bam | bedtools bamtobed -i stdin | head
chr7 118970079 118970129 TUPAC_0001:3:1:0:1452#0/1 37 -
chr7 118965072 118965122 TUPAC_0001:3:1:0:1452#0/2 37 +
chr11 46769934 46769984 TUPAC_0001:3:1:0:1472#0/1 37 -
chr11 46765606 46765656 TUPAC_0001:3:1:0:1472#0/2 37 +
chr20 54704674 54704724 TUPAC_0001:3:1:1:1833#0/1 37 +
chr20 54708987 54709037 TUPAC_0001:3:1:1:1833#0/2 37 -
chrX 9380413 9380463 TUPAC_0001:3:1:1:285#0/1 0 -
chrX 9375861 9375911 TUPAC_0001:3:1:1:285#0/2 0 +
chrX 131756978 131757028 TUPAC_0001:3:1:2:523#0/1 37 +
chrX 131761790 131761840 TUPAC_0001:3:1:2:523#0/2 37 -
-split Creating BED12 features from “spliced” BAM entries.¶
bedtools bamtobed will, by default, create a BED6 feature that represents
the entire span of a spliced/split BAM alignment. However, when using the
-split command, a BED12 feature is reported where BED blocks will be
created for each aligned portion of the sequencing read.
Chromosome ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Exons *************** **********
BED/BAM A ^^^^^^^^^^^^....................................^^^^
Result =============== ====
bamtofastq¶
bedtools bamtofastq is a conversion utility for extracting FASTQ records
from sequence alignments in BAM format.
Note
If you are using CRAM as input, you will need to specify the full path describing the location of the relevant reference genome in FASTA format via the CRAM_REFERENCE environment variable. For example:
export CRAM_REFERENCE=/path/to/ref/g1k_v37_decoy.fa
Usage and option summary¶
Usage:
bedtools bamtofastq [OPTIONS] -i <BAM> -fq <FASTQ>
(or):
bamToFastq [OPTIONS] -i <BAM> -fq <FASTQ>
| Option | Description |
|---|---|
| -fq2 | FASTQ for second end. Used if BAM contains paired-end data.
BAM should be sorted by query name
(samtools sort -n -o aln.qsort.bam aln.bam) if creating
paired FASTQ with this option. |
| -tags | Create FASTQ based on the mate info in the BAM R2 and Q2 tags. |
Default behavior¶
By default, each alignment in the BAM file is converted to a FASTQ record
in the -fq file. The order of the records in the resulting FASTQ exactly
follows the order of the records in the BAM input file.
$ bedtools bamtofastq -i NA18152.bam -fq NA18152.fq
$ head -8 NA18152.fq
@NA18152-SRR007381.35051
GGAGACATATCATATAAGTAATGCTAGGGTGAGTGGTAGGAAGTTTTTTCATAGGAGGTGTATGAGTTGGTCGTAGCGGAATCGGGGGTATGCTGTTCGAATTCATAAGAACAGGGAGGTTAGAAGTAGGGTCTTGGTGACAAAATATGTTGTATAGAGTTCAGGGGAGAGTGCGTCATATGTTGTTCCTAGGAAGATTGTAGTGGTGAGGGTGTTTATTATAATAATGTTTGTGTATTCGGCTATGAAGAATAGGGCGAAGGGGCCTGCGGCGTATTCGATGTTGAAGCCTGAGACTAGTTCGGACTCCCCTTCGGCAAGGTCGAA
+
<<<;;<;<;;<;;;;;;;;;;;;<<<:;;;;;;;;;;;;;;;;::::::;;;;<<;;;;;;;;;;;;;;;;;;;;;;;;;;;;<<<<<;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;<<;;;;;:;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;<<<;;;;;;;;;;<<<<<<<<;;;;;;;;;:;;;;;;;;;;;;;;;;;;;:;;;;8;;8888;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;8966689666666299866669:899
@NA18152-SRR007381.637219
AATGCTAGGGTGAGTGGTAGGAAGTTTTTTCATAGGAGGTGTATGAGTTGGTCGTAGCGGAATCGGGGGTATGCTGTTCGAATTCATAAGAACAGGGAGGTTAGAAGTAGGGTCTTGGTGACAAAATATGTTGTATAGAGTTCAGGGGAGAGTGCGTCATATGTTGTTCCTAGGAAGATTGTAGTGGTGAGGGTGTTTATTATAATAATGTTTGTGTATTCGGCTATGAAGAATAGGGCGAAGGGGCCTGCGGCGTATTCGATGTTGAAGCCTGAGACTAGTTCGGACTCCCCTTCCGGCAAGGTCGAA
+
<<<<<<<<<<;;<;<;;;;<<;<888888899<;;;;;;<;;;;;;;;;;;;;;;;;;;;;;;;<<<<<;;;;;;;;;<;<<<<<;;;;;;;;;;;;;<<<<;;;;;;;:::;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;<<<<;;;;;;;;;;;;;;;;;;;;;;;<;;;;;;;;;;;;;;;;;;;;;;<888<;<<;;;;<<<<<<;;;;;<<<<<<<<;;;;;;;;;:;;;;888888899:::;;8;;;;;;;;;;;;;;;;;;;99;;99666896666966666600;96666669966
-fq2 Creating two FASTQ files for paired-end sequences.¶
If your BAM alignments are from paired-end sequence data, one can use the
-fq2 option to create two distinct FASTQ output files — one for
end 1 and one for end 2.
Note
When using this option, it is required that the BAM
file is sorted/grouped by the read name. This keeps the resulting records
in the two output FASTQ files in the same order. One can sort the BAM
file by query name with samtools sort -n -o aln.qsort.bam aln.bam.
$ samtools sort -n -o aln.qsort.bam aln.bam
$ bedtools bamtofastq -i aln.qsort.bam \
-fq aln.end1.fq \
-fq2 aln.end2.fq
$ head -8 aln.end1.fq
@SRR069529.2276/1
CAGGGAGAAGGAGGTAGGAAAGAGAAAGGACCAGGGAGGGGCGCATACACAGGACGCTCCGTGCGGTGATAGCAGCACCACACTGTGTTCAGTCGTCTGGC
+
=;@>==###############################################################################################
@SRR069529.2406/1
GCTGGGAAAAGGATTCAGGATGTTGGTTTCTATCTTTGAGTTGCTGCTGTGCGGCTGTCCCTACACTCGCAGTACCCCTCGGACACCGTCTACTGTGGAGG
+
=5@><<:?<?
$ head -8 aln.end2.fq
@SRR069529.2276/2
AGACCCAGAGAGGGACAGGATCTGTCCCAGATCATAAAATAGGGGGAGTGCTCCGTAGAGGCGTGCGCGGTGGCACCGTGCAGTAGTACGGGTGAGCGGGG
+
#####################################################################################################
@SRR069529.2406/2
TTCCCTACCCCTGGGGTCAGGGACTACAGCCAAGGGGAGAACTTTAGCAAGTAGACGTTAGTTATTTTGATTCCAGTGGGGACGCGCGTGTAGCGAGTTGT
+
@>=AABB?AAACABBA>@?AAAA>B@@AB@AA:B@AA@??#############################################################
bed12tobed6¶
bed12ToBed6 is a convenience tool that converts BED features in BED12 (a.k.a. “blocked” BED features such as genes) to discrete BED6 features. For example, in the case of a gene with six exons, bed12ToBed6 would create six separate BED6 features (i.e., one for each exon).
Usage and option summary¶
Usage:
bed12ToBed6 [OPTIONS] -i <BED12>
| Option | Description |
|---|---|
| -i | The BED12 file that should be split into discrete BED6 features. Use “stdin” when using piped input. |
Default behavior¶
Figure:
head data/knownGene.hg18.chr21.bed | tail -n 3
chr21 10079666 10120808 uc002yiv.1 0 - 10081686 1 0 1 2 0 6 0 8 0 4 528,91,101,215, 0,1930,39750,40927,
chr21 10080031 10081687 uc002yiw.1 0 - 10080031 1 0 0 8 0 0 3 1 0 2 200,91, 0,1565,
chr21 10081660 10120796 uc002yix.2 0 - 10081660 1 0 0 8 1 6 6 0 0 3 27,101,223,0,37756,38913,
head data/knownGene.hg18.chr21.bed | tail -n 3 | bed12ToBed6 -i stdin
chr21 10079666 10080194 uc002yiv.1 0 -
chr21 10081596 10081687 uc002yiv.1 0 -
chr21 10119416 10119517 uc002yiv.1 0 -
chr21 10120593 10120808 uc002yiv.1 0 -
chr21 10080031 10080231 uc002yiw.1 0 -
chr21 10081596 10081687 uc002yiw.1 0 -
chr21 10081660 10081687 uc002yix.2 0 -
chr21 10119416 10119517 uc002yix.2 0 -
chr21 10120573 10120796 uc002yix.2 0 -
bedpetobam¶
bedtobam¶
bedToBam converts features in a feature file to BAM format. This is useful as an efficient means of storing large genome annotations in a compact, indexed format for visualization purposes.
Usage and option summary¶
Usage:
bedToBam [OPTIONS] -i <BED/GFF/VCF> -g <GENOME> > <BAM>
| Option | Description |
|---|---|
| -mapq | Set a mapping quality (SAM MAPQ field) value for all BED entries. Default: 255 |
| -ubam | Write uncompressed BAM output. The default is write compressed BAM output. |
| -bed12 | Indicate that the input BED file is in BED12 (a.k.a “blocked” BED) format. In this case, bedToBam will convert blocked BED features (e.g., gene annotations) into “spliced” BAM alignments by creating an appropriate CIGAR string. |
Default behavior¶
The default behavior is to assume that the input file is in unblocked format. For example:
head -5 rmsk.hg18.chr21.bed
chr21 9719768 9721892 ALR/Alpha 1004 +
chr21 9721905 9725582 ALR/Alpha 1010 +
chr21 9725582 9725977 L1PA3 3288 +
chr21 9726021 9729309 ALR/Alpha 1051 +
chr21 9729320 9729809 L1PA3 3897 -
bedToBam -i rmsk.hg18.chr21.bed -g human.hg18.genome > rmsk.hg18.chr21.bam
samtools view rmsk.hg18.chr21.bam | head -5
ALR/Alpha 0 chr21 9719769 255 2124M * 0 0 * *
ALR/Alpha 0 chr21 9721906 255 3677M * 0 0 * *
L1PA3 0 chr21 9725583 255 395M * 0 0 * *
ALR/Alpha 0 chr21 9726022 255 3288M * 0 0 * *
L1PA3 16 chr21 9729321 255 489M * 0 0 * *
Creating “spliced” BAM entries from “blocked” BED features¶
Optionally, bedToBam will create spliced BAM entries from “blocked” BED features by using the -bed12 option. This will create CIGAR strings in the BAM output that will be displayed as “spliced” alignments. The image illustrates this behavior, as the top track is a BAM representation (using bedToBam) of a BED file of UCSC genes.
For example:
bedToBam -i knownGene.hg18.chr21.bed -g human.hg18.genome -bed12 > knownGene.bam
samtools view knownGene.bam | head -2
uc002yip.1 16 chr21 9928614 2 5 5
298M1784N71M1411N93M3963N80M1927N106M3608N81M1769N62M11856N89M98N82M816N61M6910N65M
738N64M146N100M1647N120M6478N162M1485N51M6777N60M9274N54M880N54M1229N54M2377N54M112
68N58M2666N109M2885N158M * 0 0 * *
uc002yiq.1 16 chr21 9928614 2 5 5
298M1784N71M1411N93M3963N80M1927N106M3608N81M1769N62M11856N89M98N82M816N61M6910N65M
738N64M146N100M1647N120M6478N162M1485N51M6777N60M10208N54M1229N54M2377N54M11268N58M
2666N109M2885N158M * 0 0 * *
closest¶
Similar to intersect, closest searches for overlapping features in A and B. In the event that no feature in B overlaps the current feature in A, closest will report the nearest (that is, least genomic distance from the start or end of A) feature in B. For example, one might want to find which is the closest gene to a significant GWAS polymorphism. Note that closest will report an overlapping feature as the closest—that is, it does not restrict to closest non-overlapping feature. The following iconic “cheatsheet” summarizes the functionality available through the various options provided by the closest tool.

Note
bedtools closest requires that all input files are presorted data by chromosome and
then by start position (e.g., sort -k1,1 -k2,2n in.bed > in.sorted.bed
for BED files).
Note
Reports “none” for chrom and “-1” for all other fields when a feature is not found in B on the same chromosome as the feature in A. E.g. none -1 -1
Important
As of version 2.22.0, the closest tool can accept multiple files for the -b option. This allows one to identify the closest intervals between a single query (-a) file and multiple database files (-b) at once! This functionality now requires that all input files be sorted by chromosome and start coordinate in an identical manner (e.g., sort -k1,1 -k2,2n).
Usage and option summary¶
Usage:
bedtools closest [OPTIONS] -a <FILE> \
-b <FILE1, FILE2, ..., FILEN>
(or):
closestBed [OPTIONS] -a <FILE> \
-b <FILE1, FILE2, ..., FILEN>
| Option | Description |
|---|---|
| -s | Require same strandedness. That is, find the closest feature in B that overlaps A on the _same_ strand. By default, overlaps are reported without respect to strand. |
| -S | Require opposite strandedness. That is, find the closest feature in B that overlaps A on the _opposite_ strand. By default, overlaps are reported without respect to strand. |
| -d | In addition to the closest feature in B, report its distance to A as an extra column. The reported distance for overlapping features will be 0. |
| -D | Like -d, report the closest feature in B, and its distance to A as an extra column. However unlike -d, use negative distances to report upstream features.
The options for defining which orientation is “upstream” are:
- ref Report distance with respect to the reference genome.
B features with a lower (start, stop) are upstream
- a Report distance with respect to A.
When A is on the - strand, “upstream” means B has a
higher (start,stop).
- b Report distance with respect to B.
When B is on the - strand, “upstream” means A has a
higher (start,stop).
|
| -io | Ignore features in B that overlap A. That is, we want close, yet not touching features only. |
| -iu | Ignore features in B that are upstream of features in A. This option requires -D and follows its orientation rules for determining what is “upstream”. |
| -id | Ignore features in B that are downstream of features in A. This option requires -D and follows its orientation rules for determining what is “downstream”. |
| -fu | Choose first from features in B that are upstream of features in A. This option requires -D and follows its orientation rules for determining what is “upstream”. |
| -fd | Choose first from features in B that are downstream of features in A. This option requires -D and follows its orientation rules for determining what is “downstream”. |
| -t | Specify how ties for closest feature should be handled. This occurs when two features in B have exactly the same “closeness” with A. By default, all such features in B are reported.
Here are all the options:
- all Report all ties (default).
- first Report the first tie that occurred in the B file.
- last Report the last tie that occurred in the B file.
|
| -mdb | Specify how multiple databases should be resolved.
- each Report closest records for each database (default).
- all Report closest records among all databases.
|
| -k | Report the k closest hits. Default is 1. If tieMode = “all”, all ties will still be reported. |
| -names | When using multiple databases (-b), provide an alias for each that will appear instead of a fileId when also printing the DB record. |
| -filenames | When using multiple databases (-b), show each complete filename instead of a fileId when also printing the DB record. |
| -N | Require that the query and the closest hit have different names. For BED, the 4th column is compared. |
| -header | Print the header from the A file prior to results. |
Default behavior¶
The closest tool first searches for features in B that overlap a feature in A. If overlaps are found, each feature in B that overlaps A is reported. If no overlaps are found, closestBed looks for the feature in B that is closest (that is, least genomic distance to the start or end of A) to A. For example,
For example, consider the case where one of the intervals in B overlaps the interval in B, yet another does not:
$ cat a.bed
chr1 10 20 a1 1 -
$ cat b.bed
chr1 7 8 b1 1 -
chr1 15 25 b2 2 +
$ bedtools closest -a a.bed -b b.bed
chr1 10 20 a1 1 - chr1 15 25 b2 2 +
Now compare what happens when neither interval in B overlaps the record in A, yet one is closer than the other.
$ cat a.bed
chr1 10 20 a1 1 -
$ cat b.bed
chr1 7 8 b1 1 -
chr1 30 40 b2 2 +
$ bedtools closest -a a.bed -b b.bed
chr1 10 20 a1 1 - chr1 7 8 b1 1
But what if each interval in B is equally close to the interval in A? In this case, the default behavior is to report all intervals in B that are tied for proximity. Check out the -t option to adjust this behaviour.
$ cat a.bed
chr1 10 20 a1 1 -
$ cat b.bed
chr1 7 8 b1 1 -
chr1 22 23 b2 2 +
$ bedtools closest -a a.bed -b b.bed
chr1 10 20 a1 1 - chr1 7 8 b1 1 -
chr1 10 20 a1 1 - chr1 22 23 b2 2 +
Using multiple -b files.¶
As of version, 2.22.0, the closest tool allows one to find the closest intervals in multiple -b files. Consider the following examples.
Note
When using multiple -b files, an additional column describing the file number from which the closest B interval came will be added between the columns representing the full A interval and the columns representing the full A interval. This file number will refer to the order in which the files were provided on the command line.
$ cat a.bed
chr1 10 20 a1 1 -
$ cat b1.bed
chr1 5 6 b1.1 1 -
chr1 30 40 b1.2 2 +
$ cat b2.bed
chr1 0 1 b2.1 1 -
chr1 21 22 b2.2 2 +
# In this example, the 7th column reflects the file number from
# which the closest interval came.
$ bedtools closest -a a.bed -b b1.bed b2.bed
chr1 10 20 a1 1 - 1 chr1 5 6 b1.1 1 -
chr1 10 20 a1 1 - 2 chr1 21 22 b2.2 2 +
Instead of using file numbers, you can also provide more informative labels via the -names option.
$ bedtools closest -a a.bed -b b1.bed b2.bed -names b1 b2
chr1 10 20 a1 1 - b1 chr1 5 6 b1.1 1 -
chr1 10 20 a1 1 - b2 chr1 21 22 b2.2 2 +
Or, you can use the full original filename via the -filenames option.
$ bedtools closest -a a.bed -b b1.bed b2.bed -filenames
chr1 10 20 a1 1 - b1.bed chr1 5 6 b1.1 1 -
chr1 10 20 a1 1 - b2.bed chr1 21 22 b2.2 2 +
-mdb Find the closest interval in each* or among **all -b files.¶
By default, the closest interval from each file is reported when using multiple -b files.
$ cat a.bed
chr1 10 20 a1 1 -
$ cat b1.bed
chr1 5 6 b1.1 1 -
chr1 30 40 b1.2 2 +
$ cat b2.bed
chr1 0 1 b2.1 1 -
chr1 21 22 b2.2 2 +
$ bedtools closest -a a.bed -b b1.bed b2.bed -d
chr1 10 20 a1 1 - 1 chr1 5 6 b1.1 1 - 5
chr1 10 20 a1 1 - 2 chr1 21 22 b2.2 2 + 2
$ bedtools closest -a a.bed -b b1.bed b2.bed -mdb each -d
chr1 10 20 a1 1 - 1 chr1 5 6 b1.1 1 - 5
chr1 10 20 a1 1 - 2 chr1 21 22 b2.2 2 + 2
However, one can optionally choose to report only the closest interval(s) observed among all of the -b files. In this example, the second interval from b2.bed is only 2 base pairs away from the interval in A, whereas the first interval in b1.bed is 5 base pairs away. Therefore, when using mdb all, the the second interval from b2.bed wins.
$ bedtools closest -a a.bed -b b1.bed b2.bed -mdb all -d
chr1 10 20 a1 1 - 2 chr1 21 22 b2.2 2 + 2
-io Ignoring overlapping intervals¶
This option prevents intervals in B that overlap the interval in A from being reported as “closest”.
Without -ip the second record in B will be reported as closest.
$ cat a.bed
chr1 10 20 a1 1 -
$ cat b.bed
chr1 7 8 b1 1 -
chr1 15 25 b2 2 +
$ bedtools closest -a a.bed -b b.bed
chr1 10 20 a1 1 - chr1 15 25 b2 2 +
Yet with -io, the overlapping interval is ignored in favor of the closest, non-overlapping interval.
$ cat a.bed
chr1 10 20 a1 1 -
$ cat b.bed
chr1 7 8 b1 1 -
chr1 15 25 b2 2 +
$ bedtools closest -a a.bed -b b.bed -io
chr1 10 20 a1 1 - chr1 7 8 b1 1 -
-s Requiring closest intervals to have the same strand¶
The -s option finds the closest interval that is also on the same strand as the interval in A.
$ cat a.bed
chr1 10 20 a1 1 -
$ cat b.bed
chr1 2 3 b1 1 -
chr1 21 22 b2 2 +
$ bedtools closest -a a.bed -b b.bed -s
chr1 10 20 a1 1 - chr1 2 3 b1 1 -
-S Requiring closest intervals to have the opposite strand¶
The -s option finds the closest interval that is also on the same strand as the interval in A.
$ cat a.bed
chr1 10 20 a1 1 -
$ cat b.bed
chr1 15 16 b1 1 -
chr1 21 22 b2 2 +
$ bedtools closest -a a.bed -b b.bed -S
chr1 10 20 a1 1 - chr1 21 22 b2 2 +
-t Controlling how ties for “closest” are broken¶
When there are two or more features in B are tied for proximity to the interval in A, closest will, by default, report all such intervals in B. As shown in the examples below, this behavior can be changed via the -t option:
$ cat a.bed
chr1 10 20 a1 1 -
$ cat b.bed
chr1 30 40 b1 1 -
chr1 30 40 b2 2 +
# default
$ bedtools closest -a a.bed -b b.bed
chr1 10 20 a1 1 - chr1 30 40 b1 1 -
chr1 10 20 a1 1 - chr1 30 40 b2 2 +
# -t all (default)
$ bedtools closest -a a.bed -b b.bed -t all
chr1 10 20 a1 1 - chr1 30 40 b1 1 -
chr1 10 20 a1 1 - chr1 30 40 b2 2 +
# -t first
$ bedtools closest -a a.bed -b b.bed -t first
chr1 10 20 a1 1 - chr1 30 40 b1 1 -
# -t last
$ bedtools closest -a a.bed -b b.bed -t last
chr1 10 20 a1 1 - chr1 30 40 b2 1 +
-d Reporting the distance to the closest feature in base pairs¶
One often wants to also know the distance in base pairs between the interval in A and the closest interval(s) in B. closest will optionally report the distance to the closest feature in the B file using the -d option. The distance (in base pairs) will be reported as the last column in the output.
When a feature in B overlaps a feature in A, a distance of 0 is reported.
$ cat a.bed
chr1 10 20 a1 1 -
$ cat b.bed
chr1 7 8 b1 1 -
chr1 22 23 b2 2 +
$ bedtools closest -a a.bed -b b.bed
chr1 10 20 a1 1 - chr1 7 8 b1 1 - 3
chr1 10 20 a1 1 - chr1 22 23 b2 2 + 3
-D Reporting signed distances to the closest feature in base pairs¶
Whereas the -d option always reports distances as positive integers, the -D option will use negative integers to report distances to “upstream” features. There are three options for dictating how “upstream” should be defined.
- -D ref: Report distance with respect to the reference genome. That is, B features with lower start/stop coordinates are considered to be upstream.
- -D a: Report distance with respect to the orientation of the interval in A. That is, when A is on the - strand, “upstream” means B has higher start/stop coordinates. When A is on the + strand, “upstream” means B has lower start/stop coordinates.
- -D b: Report distance with respect to the orientation of the interval in B. That is, when B is on the - strand, “upstream” means A has higher start/stop coordinates. When B is on the + strand, “upstream” means A has lower start/stop coordinates.
This is best demonstrated through multiple examples.
$ cat a.bed
chr1 10 20 a1 1 +
$ cat b.bed
chr1 7 8 b1 1 +
chr1 22 23 b2 2 -
$ bedtools closest -a a.bed -b b.bed -D ref
chr1 10 20 a1 1 + chr1 7 8 b1 1 + -3
chr1 10 20 a1 1 + chr1 22 23 b2 2 - 3
Since the A record is on the “+” strand in this example, -D ref and -D a have the same effect.
$ bedtools closest -a a.bed -b b.bed -D a
chr1 10 20 a1 1 + chr1 7 8 b1 1 + -3
chr1 10 20 a1 1 + chr1 22 23 b2 2 - 3
However, the signs of the distances change if the A interval is on the “-” strand.
$ cat a.bed
chr1 10 20 a1 1 -
$ bedtools closest -a a.bed -b b.bed -D a
chr1 10 20 a1 1 - chr1 7 8 b1 1 + 3
chr1 10 20 a1 1 - chr1 22 23 b2 2 - -3
Let’s switch the A interval back to the “+” strand and now report distances with respect to the orientation of the closest B records.
$ cat a.bed
chr1 10 20 a1 1 +
$ bedtools closest -a a.bed -b b.bed -D b
chr1 10 20 a1 1 + chr1 7 8 b1 1 + 3
chr1 10 20 a1 1 + chr1 22 23 b2 2 - 3
Let’s flip the stand of the two B records and compare.
$ cat b.bed
chr1 7 8 b1 1 -
chr1 22 23 b2 2 +
$ bedtools closest -a a.bed -b b.bed -D b
chr1 10 20 a1 1 + chr1 7 8 b1 1 - -3
chr1 10 20 a1 1 + chr1 22 23 b2 2 + -3
cluster¶
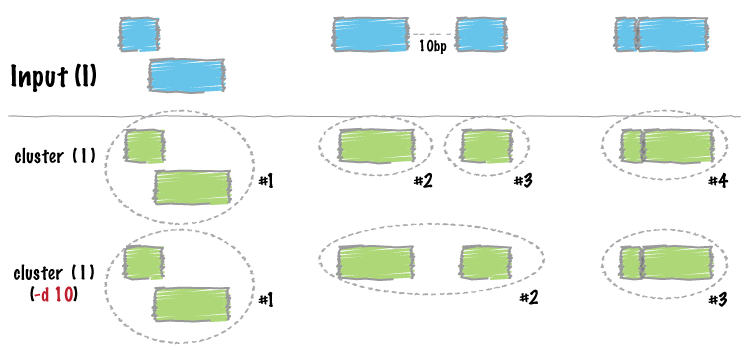
Similar to merge, cluster report each set of overlapping or
“book-ended” features in an interval file. In contrast to merge,
cluster does not flatten the cluster of intervals into a new meta-interval;
instead, it assigns an unique cluster ID to each record in each cluster. This
is useful for having fine control over how sets of overlapping intervals in
a single interval file are combined.
Note
bedtools cluster requires that you presort your data by chromosome and
then by start position (e.g., sort -k1,1 -k2,2n in.bed > in.sorted.bed
for BED files).
See also
Usage and option summary¶
Usage:
bedtools cluster [OPTIONS] -i <BED/GFF/VCF>
(or):
clusterBed [OPTIONS] -i <BED/GFF/VCF>
| Option | Description |
|---|---|
| -s | Force strandedness. That is, only cluster features that are the same strand. By default, this is disabled. |
| -d | Maximum distance between features allowed for features to be clustered. Default is 0. That is, overlapping and/or book-ended features are clustered. |
Default behavior¶
By default, bedtools cluster collects overlapping (by at least 1 bp) and/or
bookended intervals into distinct clusters. In the example below, the 4th
column is the cluster ID.
$ cat A.bed
chr1 100 200
chr1 180 250
chr1 250 500
chr1 501 1000
$ bedtools cluster -i A.bed
chr1 100 200 1
chr1 180 250 1
chr1 250 500 1
chr1 501 1000 2
-s Enforcing “strandedness”¶
The -s option will only cluster intervals that are overlapping/bookended
and are on the same strand.
$ cat A.bed
chr1 100 200 a1 1 +
chr1 180 250 a2 2 +
chr1 250 500 a3 3 -
chr1 501 1000 a4 4 +
$ bedtools cluster -i A.bed -s
chr1 100 200 a1 1 + 1
chr1 180 250 a2 2 + 1
chr1 501 1000 a4 4 + 2
chr1 250 500 a3 3 - 3
-d Controlling how close two features must be in order to cluster¶
By default, only overlapping or book-ended features are combined into a new
feature. However, one can force cluster to combine more distant features
with the -d option. For example, were one to set -d to 1000, any
features that overlap or are within 1000 base pairs of one another will be
clustered.
$ cat A.bed
chr1 100 200
chr1 501 1000
$ bedtools cluster -i A.bed
chr1 100 200 1
chr1 501 1000 2
$ bedtools cluster -i A.bed -d 1000
chr1 100 200 1
chr1 501 1000 1
complement¶
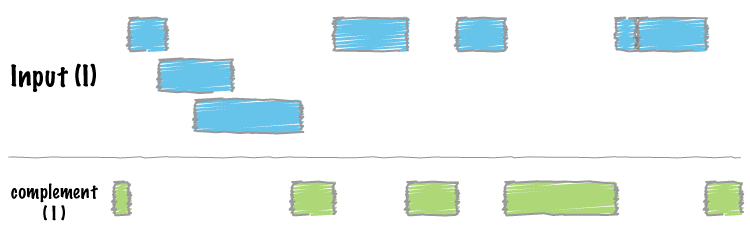
bedtools complement returns all intervals in a genome that are not
covered by at least one interval in the input BED/GFF/VCF file.
See also
Usage and option summary¶
Usage:
bedtools complement -i <BED/GFF/VCF> -g <GENOME>
(or):
complementBed -i <BED/GFF/VCF> -g <GENOME>
Default behavior¶
By default, bedtools complement returns all genomic intervals that are not
covered by at least one record from the input file.
$ cat A.bed
chr1 100 200
chr1 400 500
chr1 500 800
$ cat my.genome
chr1 1000
chr2 800
$ bedtools complement -i A.bed -g my.genome
chr1 0 100
chr1 200 400
chr1 800 1000
chr2 0 800
-L Only report chromosomes that are in the -i file¶
Use the “-L” option to L`imit the output to solely the chromosomes that are represented in the `-i file. Chromosomes that are in -g but not -i will be suppressed
For example (note the difference in coverage with and without -s:
$ cat A.bed
chr1 100 200
chr1 400 500
chr1 500 800
$ cat my.genome
chr1 1000
chr2 800
$ bedtools complement -i A.bed -g my.genome
chr1 0 100
chr1 200 400
chr1 800 1000
coverage¶
The bedtools coverage tool computes both the depth and breadth of coverage of features in file B on the features
in file A. For example, bedtools coverage can compute the coverage of sequence alignments (file B) across 1
kilobase (arbitrary) windows (file A) tiling a genome of interest. One advantage that bedtools coverage
offers is that it not only counts the number of features that overlap an interval in file A, it also
computes the fraction of bases in the interval in A that were overlapped by one or more features. Thus,
bedtools coverage also computes the breadth of coverage observed for each interval in A.
Note
If you are trying to compute coverage for very large files and are having trouble
with excessive memory usage, please presort your data by chromosome and
then by start position (e.g., sort -k1,1 -k2,2n in.bed > in.sorted.bed
for BED files) and then use the -sorted option. This invokes a
memory-efficient algorithm designed for large files.
Important
As of version 2.24.0, the coverage tool has changed such that the coverage is computed for the A file, not the B file. This changes the command line interface to be consistent with the other tools. Also, the coverage tool can accept multiple files for the -b option. This allows one to measure coverage between a single query (-a) file and multiple database files (-b) at once!
Usage and option summary¶
Usage:
bedtools coverage [OPTIONS] -a <FILE> \
-b <FILE1, FILE2, ..., FILEN>
(or):
coverageBed [OPTIONS] -a <FILE> \
-b <FILE1, FILE2, ..., FILEN>
| Option | Description |
|---|---|
| -a | BAM/BED/GFF/VCF file “A”. Each feature in A is compared to B in search of overlaps. Use “stdin” if passing A with a UNIX pipe. |
| -b | One or more BAM/BED/GFF/VCF file(s) “B”. Use “stdin” if passing B with a UNIX pipe. NEW!!!: -b may be followed with multiple databases and/or wildcard (*) character(s). |
| -abam | BAM file A. Each BAM alignment in A is compared to B in search of overlaps. Use “stdin” if passing A with a UNIX pipe: For example: samtools view -b <BAM> | bedtools intersect -abam stdin -b genes.bed. Note: no longer necessary after version 2.19.0 |
| -hist | Report a histogram of coverage for each feature in A as well as a summary histogram for _all_ features in A.
Output (tab delimited) after each feature in A:
1) depth
2) # bases at depth
3) size of A
4) % of A at depth
|
| -d | Report the depth at each position in each A feature. Positions reported are one based. Each position and depth follow the complete A feature. |
| -counts | Only report the count of overlaps, don’t compute fraction, etc. Restricted by -f and -r. |
| -f | Minimum overlap required as a fraction of A. Default is 1E-9 (i.e. 1bp). |
| -F | Minimum overlap required as a fraction of B. Default is 1E-9 (i.e., 1bp). |
| -r | Require that the fraction of overlap be reciprocal for A and B. In other words, if -f is 0.90 and -r is used, this requires that B overlap at least 90% of A and that A also overlaps at least 90% of B. |
| -e | Require that the minimum fraction be satisfied for A _OR_ B. In other words, if -e is used with -f 0.90 and -F 0.10 this requires that either 90% of A is covered OR 10% of B is covered. Without -e, both fractions would have to be satisfied. |
| -s | Force “strandedness”. That is, only report hits in B that overlap A on the same strand. By default, overlaps are reported without respect to strand. |
| -S | Require different strandedness. That is, only report hits in B that overlap A on the _opposite_ strand. By default, overlaps are reported without respect to strand. |
| -split | Treat “split” BAM (i.e., having an “N” CIGAR operation) or BED12 entries as distinct BED intervals. |
| -sorted | For very large B files, invoke a “sweeping” algorithm that requires position-sorted (e.g., sort -k1,1 -k2,2n for BED files) input. When using -sorted, memory usage remains low even for very large files. |
| -g | Specify a genome file the defines the expected chromosome order in the input files for use with the -sorted option. |
| -header | Print the header from the A file prior to results. |
| -sortout | When using multiple databases (-b), sort the output DB hits for each record. |
| -nobuf | Disable buffered output. Using this option will cause each line of output to be printed as it is generated, rather than saved in a buffer. This will make printing large output files noticeably slower, but can be useful in conjunction with other software tools and scripts that need to process one line of bedtools output at a time. |
| -iobuf | Follow with desired integer size of read buffer. Optional suffixes K/M/G supported. Note: currently has no effect with compressed files. |
Default behavior¶
After each interval in A, bedtools coverage will report:
- The number of features in B that overlapped (by at least one base pair) the A interval.
- The number of bases in A that had non-zero coverage from features in B.
- The length of the entry in A.
- The fraction of bases in A that had non-zero coverage from features in B.
Below are the number of features in B (N=…) overlapping A and fraction of bases in A with coverage.
Chromosome ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
BED FILE A *************** *************** ****** **************
BED File B ^^^^ ^^^^ ^^ ^^^^^^^^^ ^^^ ^^ ^^^^
^^^^^^^^ ^^^^^ ^^^^^ ^^
Result [ N=3, 10/15 ] [ N=1, 2/15 ] [N=1,6/6] [N=6, 12/14 ]
For example:
$ cat A.bed
chr1 0 100
chr1 100 200
chr2 0 100
$ cat B.bed
chr1 10 20
chr1 20 30
chr1 30 40
chr1 100 200
$ bedtools coverage -a A.bed -b B.bed
chr1 0 100 3 30 100 0.3000000
chr1 100 200 1 100 100 1.0000000
chr2 0 100 0 0 100 0.0000000
-s Calculating coverage by strand¶
Use the “-s” option if one wants to only count coverage if features in A are on the same strand as the feature / window in A. This is especially useful for RNA-seq experiments.
For example (note the difference in coverage with and without -s:
$ cat A.bed
chr1 0 100 b1 1 +
chr1 100 200 b2 1 -
chr2 0 100 b3 1 +
$ cat B.bed
chr1 10 20 a1 1 -
chr1 20 30 a2 1 -
chr1 30 40 a3 1 -
chr1 100 200 a4 1 +
$ bedtools coverage -a A.bed -b B.bed
chr1 0 100 b1 1 + 3 30 100 0.3000000
chr1 100 200 b2 1 - 1 100 100 1.0000000
chr2 0 100 b3 1 + 0 0 100 0.0000000
$ bedtools coverage -a A.bed -b B.bed -s
chr1 0 100 b1 1 + 0 0 100 0.0000000
chr1 100 200 b2 1 - 0 0 100 0.0000000
chr2 0 100 b3 1 + 0 0 100 0.0000000
-hist Creating a histogram of coverage for each feature in the A file¶
One should use the “-hist” option to create, for each interval in A, a histogram of coverage of the features in B across A.
In this case, each entire feature in A will be reported, followed by the depth of coverage, the number of bases at that depth, the size of the feature, and the fraction covered. After all of the features in A have been reported, a histogram summarizing the coverage among all features in A will be reported.
$ cat A.bed
chr1 0 100 b1 1 +
chr1 100 200 b2 1 -
chr2 0 100 b3 1 +
$ cat B.bed
chr1 10 20 a1 1 -
chr1 20 30 a2 1 -
chr1 30 40 a3 1 -
chr1 100 200 a4 1 +
$ bedtools coverage -a A.bed -b B.bed -hist
chr1 0 100 b1 1 + 0 70 100 0.7000000
chr1 0 100 b1 1 + 1 30 100 0.3000000
chr1 100 200 b2 1 - 1 100 100 1.0000000
chr2 0 100 b3 1 + 0 100 100 1.0000000
all 0 170 300 0.5666667
all 1 130 300 0.4333333
-d Reporting the per-base of coverage for each feature in the A file¶
One should use the “-d” option to create, for each interval in A, a detailed list of coverage at each of the positions across each A interval.
The output will consist of a line for each one-based position in each A feature, followed by the coverage detected at that position.
$ cat A.bed
chr1 0 10
$ cat B.bed
chr1 0 5
chr1 3 8
chr1 4 8
chr1 5 9
$ bedtools coverage -a A.bed -b B.bed -d
chr1 0 10 B 1 1
chr1 0 10 B 2 1
chr1 0 10 B 3 1
chr1 0 10 B 4 2
chr1 0 10 B 5 3
chr1 0 10 B 6 3
chr1 0 10 B 7 3
chr1 0 10 B 8 3
chr1 0 10 B 9 1
chr1 0 10 B 10 0
expand¶
flank¶
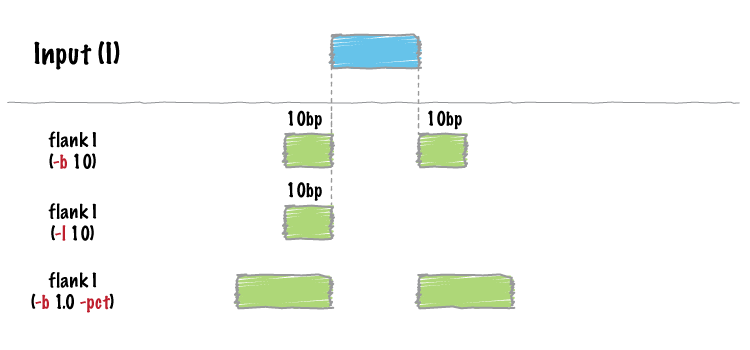
bedtools flank will create two new flanking intervals for each interval in
a BED/GFF/VCF file. Note that flank will restrict the created flanking
intervals to the size of the chromosome (i.e. no start < 0 and no end >
chromosome size).
Note
In order to prevent creating intervals that violate chromosome boundaries,
bedtools flank requires a genome file defining the length of each
chromosome or contig.
See also
Usage and option summary¶
Usage:
bedtools flank [OPTIONS] -i <BED/GFF/VCF> -g <GENOME> [-b or (-l and -r)]
(or):
flankBed [OPTIONS] -i <BED/GFF/VCF> -g <GENOME> [-b or (-l and -r)]
| Option | Description |
|---|---|
| -b | Increase the BED/GFF/VCF entry by the same number base pairs in each direction. Integer. |
| -l | The number of base pairs to subtract from the start coordinate. Integer. |
| -r | The number of base pairs to add to the end coordinate. Integer. |
| -s | Define -l and -r based on strand. For example. if used, -l 500 for a negative-stranded feature, it will add 500 bp to the end coordinate. |
| -pct | Define -l and -r as a fraction of the feature’s length. E.g. if used on a 1000bp feature, -l 0.50, will add 500 bp “upstream”. Default = false. |
Default behavior¶
By default, bedtools flank will either add a fixed number of bases in each
direction (-b) or an asymmetric number of bases in each direction
with -l and -r.
$ cat A.bed
chr1 100 200
chr1 500 600
$ cat my.genome
chr1 1000
$ bedtools flank -i A.bed -g my.genome -b 5
chr1 95 100
chr1 200 205
chr1 495 500
chr1 600 605
$ bedtools flank -i A.bed -g my.genome -l 2 -r 3
chr1 98 100
chr1 200 203
chr1 498 500
chr1 600 603
However, if the requested number of bases exceeds the boundaries of the
chromosome, bedtools flank will “clip” the feature accordingly.
$ cat A.bed
chr1 100 200
chr1 500 600
$ cat my.genome
chr1 1000
$ bedtools flank -i A.bed -g my.genome -b 800
chr1 0 100
chr1 200 1000
chr1 0 500
chr1 600 1000
-pct Resizing features by a given fraction¶
bedtools flank will optionally create flanking intervals whose size is
user-specified fraction of the original interval.
For example:
$ cat A.bed
chr1 100 200
chr1 500 700
################################################################
# note the flanking intervals from the second record in A.bed
# are 20bp whereas the flanking intervals from the first record
# are only 10bp
################################################################
$ bedtools flank -i A.bed -g my.genome -b 0.1 -pct
chr1 90 100
chr1 200 210
chr1 480 500
chr1 700 720
fisher¶
Perform fisher’s exact test on the number of overlaps/unique intervals between 2 files.
Traditionally, in order to test whether 2 sets of intervals are related spatially, we resort to shuffling the genome and checking the simulated (shuffled) versus the observed. We can do the same analytically for many scenarios using Fisher’s Exact Test .
This implementation can calculate the number of overlaps and the number of intervals unique to each file and it infers (or accepts) the number that are not present in each file.
Given a pair of input files -a and -b in the usual BedTools parlance:
$ cat a.bed
chr1 10 20
chr1 30 40
chr1 51 52
$ cat b.bed
chr1 15 25
chr1 51 52
And a genome of 500 bases:
$ echo -e "chr1\t500" > t.genome
We may wish to know if the amount of overlap between the 2 sets of intervals is
more than we would expect given their coverage and the size of the genome. We
can do this with fisher as:
$ bedtools fisher -a a.bed -b b.bed -g t.genome
# Number of query intervals: 3
# Number of db intervals: 2
# Number of overlaps: 2
# Number of possible intervals (estimated): 37
# phyper(2 - 1, 3, 37 - 3, 2, lower.tail=F)
# Contingency Table Of Counts
#_________________________________________
# | in -b | not in -b |
# in -a | 2 | 1 |
# not in -a | 0 | 34 |
#_________________________________________
# p-values for fisher's exact test
left right two-tail ratio
1 0.0045045 0.0045045 inf
Where we can see the constructed contingency table and the pvalues for left, right and two-tail tests. From here, we can say that given 500 bases of genome, it is unlikely that we would see as many overlaps as we do if the intervals from a and b were not related.
Note
the total number of possible intervals in the above example was estimated to be 37. This is based on a heuristic that uses the mean sizes of intervals in the a and b sets and the size of the genome. The reported p-value will depend greatly on this. Below, we show how well the reported value matches with simulations.
The above had a fairly low p-value (0.0045), but if our genome were only 60 bases:
$ echo -e "chr1\t60" > t.genome
$ bedtools fisher -a a.bed -b b.bed -g t.genome
# Number of query intervals: 3
# Number of db intervals: 2
# Number of overlaps: 2
# Number of possible intervals (estimated): 4
# phyper(2 - 1, 3, 4 - 3, 2, lower.tail=F)
# Contingency Table Of Counts
#_________________________________________
# | in -b | not in -b |
# in -a | 2 | 1 |
# not in -a | 0 | 1 |
#_________________________________________
# p-values for fisher's exact test
left right two-tail ratio
1 0.5 1 inf
We can see that neither tail is significant. Intuitively, this makes sense; if we randomly place 3 intervals (from -a), and 2 (from -b) intervals from within 60 bases, it doesn’t seem unlikely that we’d see 2 overlaps.
Note also that since the genome size is much smaller, the number of possible intervals is also decreased.
Evaluation¶
The p-value depends on knowing or inferring the total number of possible intervals (to fill in the lower right corner of the contingency table). This inference is not straightforward since we will most likely have variable sized intervals within and between files. Below, we show the correspondence of the p-value reported by fisher and one from simulated data.
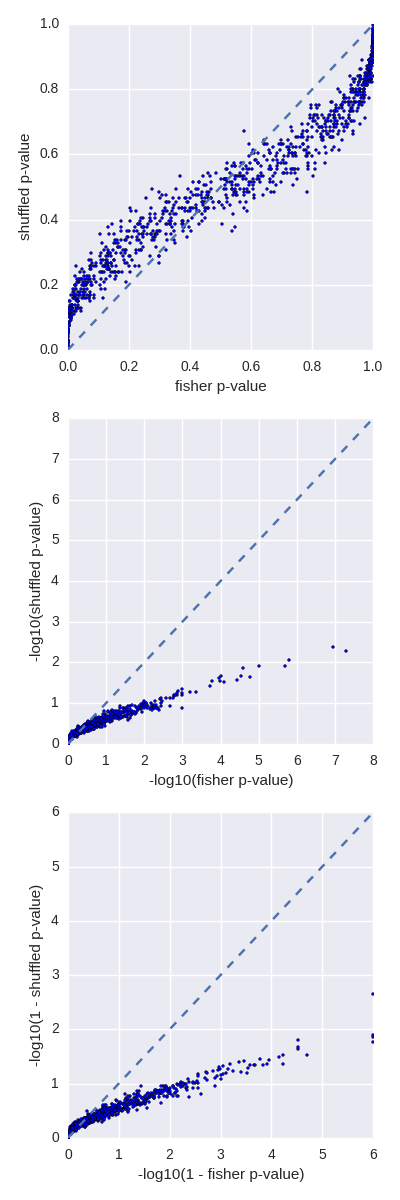
The comparison of the p-value from ‘fisher’ to one derived by simulation (see tests/fisher/ for details). The top plot shows the p-value distribution. Since we are most interested in extreme p-values, the middle plot shows -log10(p-value). The bottom plot is the same as the middle except looking at the other tail of the p-value distribution.
Note that we do see inflation from the fisher test, but we do not see ‘false-negatives’–that is, bedtools fisher is less likely to miss ‘true’ candidates, but it will give many candidates for further exploration. As such we recommend validating low p-values from fisher using simulation.
This evaluation used all known canonical genes on chromosome 1 and repeatedly (1000 times) randomly generated 3000 intervals between 20 and 5250 bases. It then calculated the p-value for each set using fisher and then using shuffled data.
Note
The fisher tool requires that your data is pre-sorted by chromosome and
then by start position (e.g., sort -k1,1 -k2,2n in.bed > in.sorted.bed
for BED files).
This uses Heng Li’s implementation of Fisher’s exact test in kfunc.c.
Usage and option summary¶
Usage:
bedtools fisher [OPTIONS] -a <BED/GFF/VCF> -b <BED/GFF/VCF> -g <genome>
| Option | Description |
|---|---|
| -a | BED/GFF/VCF file A. Each feature in A is compared to B in search of overlaps. Use “stdin” if passing A with a UNIX pipe. |
| -b | BED/GFF/VCF file B. Use “stdin” if passing B with a UNIX pipe. |
| -g | genome file listing chromosome size. |
| -f | Minimum overlap required as a fraction of A. Default is 1E-9 (i.e. 1bp). |
| -r | Require that the fraction of overlap be reciprocal for A and B. In other words, if -f is 0.90 and -r is used, this requires that B overlap at least 90% of A and that A also overlaps at least 90% of B. |
| -s | Force “strandedness”. That is, only report hits in B that overlap A on the same strand. By default, overlaps are reported without respect to strand. |
| -S | Require different strandedness. That is, only report hits in B that overlap A on the _opposite_ strand. By default, overlaps are reported without respect to strand. |
| -split | Treat “split” BAM (i.e., having an “N” CIGAR operation) or BED12 entries as distinct BED intervals. |
genomecov¶
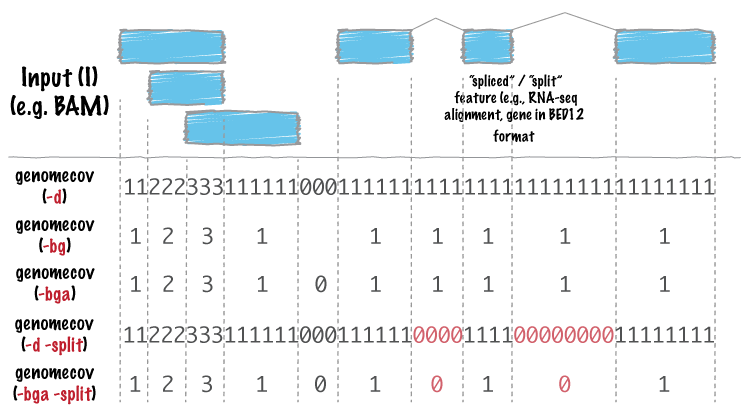
bedtools genomecov computes histograms (default), per-base reports (-d)
and BEDGRAPH (-bg) summaries of feature coverage (e.g., aligned sequences)
for a given genome.
Note
1. If using BED/GFF/VCF, the input (-i) file must be grouped by
chromosome. A simple sort -k 1,1 in.bed > in.sorted.bed will suffice.
Also, if using BED/GFF/VCF, one must provide a genome file via the -g
argument.
2. If the input is in BAM (-ibam) format, the BAM file must be sorted
by position. Using samtools sort aln.bam aln.sorted will suffice.
Usage and option summary¶
Usage:
bedtools genomecov [OPTIONS] [-i|-ibam] -g (iff. -i and not -ibam)
(or):
genomeCoverageBed [OPTIONS] [-i|-ibam] -g (iff. -i and not -ibam)
| Option | Description |
|---|---|
| -ibam | BAM file as input for coverage. Each BAM alignment in A added to the total coverage for the genome.
Use “stdin” or simply “-” if passing it with a UNIX pipe: For example:
samtools view -b <BAM> | genomeCoverageBed -ibam stdin |
| -g | Provide a genome file to define chromosome lengths. Required when not using -ibam option. |
| -d | Report the depth at each genome position with 1-based coordinates. |
| -dz | Report the depth at each genome position with 0-based coordinates. Unlike, -d, this reports only non-zero positions. |
| -bg | Report depth in BedGraph format. For details, see: http://genome.ucsc.edu/goldenPath/help/bedgraph.html |
| -bga | Report depth in BedGraph format, as above (i.e., -bg). However with this option, regions with zero coverage are also reported. This allows one to quickly extract all regions of a genome with 0 coverage by applying: “grep -w 0$” to the output. |
| -split | Treat “split” BAM or BED12 entries as distinct BED intervals when computing coverage. For BAM files, this uses the CIGAR “N” and “D” operations to infer the blocks for computing coverage. For BED12 files, this uses the BlockCount, BlockStarts, and BlockEnds fields (i.e., columns 10,11,12). |
| -strand | Calculate coverage of intervals from a specific strand. With BED files, requires at least 6 columns (strand is column 6). |
| -5 | Calculate coverage of 5’ positions (instead of entire interval). |
| -3 | Calculate coverage of 3’ positions (instead of entire interval). |
| -max | Combine all positions with a depth >= max into a single bin in the histogram. |
| -scale | Scale the coverage by a constant factor.
Each coverage value is multiplied by this factor before being reported.
Useful for normalizing coverage by, e.g., reads per million (RPM).
Default is 1.0; i.e., unscaled. |
| -trackline | Adds a UCSC/Genome-Browser track line definition in the first line of the output.
See here for more details about track line definition:
|
| -trackopts | Writes additional track line definition parameters in the first line. |
| -pc | Calculates coverage of intervals from left point of a pair reads to the right point.
Works for BAM files only
|
| -fs | Forces to use fragment size instead of read length
Works for BAM files only
|
Default behavior¶
By default, bedtools genomecov will compute a histogram of coverage for
the genome file provided. The default output format is as follows:
- chromosome (or entire genome)
- depth of coverage from features in input file
- number of bases on chromosome (or genome) with depth equal to column 2.
- size of chromosome (or entire genome) in base pairs
- fraction of bases on chromosome (or entire genome) with depth equal to column 2.
For example:
$ cat A.bed
chr1 10 20
chr1 20 30
chr2 0 500
$ cat my.genome
chr1 1000
chr2 500
$ bedtools genomecov -i A.bed -g my.genome
chr1 0 980 1000 0.98
chr1 1 20 1000 0.02
chr2 1 500 500 1
genome 0 980 1500 0.653333
genome 1 520 1500 0.346667
-max Controlling the histogram’s maximum depth¶
Using the -max option, bedtools genomecov will “lump” all positions in
the genome having feature coverage greater than or equal to -max into
the -max histogram bin. For example, if one sets -max
equal to 50, the max depth reported in the output will be 50 and all positions
with a depth >= 50 will be represented in bin 50.
-d Reporting “per-base” genome coverage¶
Using the -d option, bedtools genomecov will compute the depth of
feature coverage for each base on each chromosome in genome file provided.
The “per-base” output format is as follows:
- chromosome
- chromosome position
- depth (number) of features overlapping this chromosome position.
For example:
$ cat A.bed
chr1 10 20
chr1 20 30
chr2 0 500
$ cat my.genome
chr1 1000
chr2 500
$ bedtools genomecov -i A.bed -g my.genome -d | \
head -15 | \
tail -n 10
chr1 6 0
chr1 7 0
chr1 8 0
chr1 9 0
chr1 10 0
chr1 11 1
chr1 12 1
chr1 13 1
chr1 14 1
chr1 15 1
-bg Reporting genome coverage in BEDGRAPH format.¶
Whereas the -d option reports an output line describing the observed
coverage at each and every position in the genome, the -bg option instead
produces genome-wide coverage output in
BEDGRAPH format.
This is a much more concise representation since consecutive positions with the
same coverage are reported as a single output line describing the start and end
coordinate of the interval having the coverage level, followed by the coverage
level itself.
For example, below is a snippet of BEDGRAPH output of the coverage from a 1000 Genome Project BAM file:
$ bedtools genomecov -ibam NA18152.bam -bg | head
chr1 554304 554309 5
chr1 554309 554313 6
chr1 554313 554314 1
chr1 554315 554316 6
chr1 554316 554317 5
chr1 554317 554318 1
chr1 554318 554319 2
chr1 554319 554321 6
chr1 554321 554323 1
chr1 554323 554334 7
Using this format, one can quickly identify regions of the genome with
sufficient coverage (in this case, 10 or more reads) by piping the
output to an awk filter.
$ bedtools genomecov -ibam NA18152.bam -bg | \
awk '$4 > 9' | \
head
chr1 554377 554381 11
chr1 554381 554385 12
chr1 554385 554392 16
chr1 554392 554408 17
chr1 554408 554410 19
chr1 554410 554422 20
chr1 554422 554423 19
chr1 554423 554430 22
chr1 554430 554440 24
chr1 554440 554443 25
-bga Reporting genome coverage for all positions in BEDGRAPH format.¶
The -bg option reports coverage in BEDGRAPH format only for those regions
of the genome that actually have coverage. But what about the uncovered portion
of the genome? By using the -bga option, one receives a complete report
including the regions with zero coverage.
For example, compare the output from -bg:
$ bedtools genomecov -ibam NA18152.bam -bg | head
chr1 554304 554309 5
chr1 554309 554313 6
chr1 554313 554314 1
chr1 554315 554316 6
chr1 554316 554317 5
chr1 554317 554318 1
chr1 554318 554319 2
chr1 554319 554321 6
chr1 554321 554323 1
chr1 554323 554334 7
to the output from -bga:
# Note the first record reports that the first 554304
# base pairs of chr1 had zero coverage
$ bedtools genomecov -ibam NA18152.bam -bga | head
chr1 0 554304 0
chr1 554304 554309 5
chr1 554309 554313 6
chr1 554313 554314 1
chr1 554314 554315 0
chr1 554315 554316 6
chr1 554316 554317 5
chr1 554317 554318 1
chr1 554318 554319 2
chr1 554319 554321 6
-strand Reporting genome coverage for a specific strand.¶
Whereas the default is to count coverage regardless of strand, the -strand
option allows one to report the coverage observed for a specific strand.
Compare:
$ bedtools genomecov -ibam NA18152.bam -bg | head
chr1 554304 554309 5
chr1 554309 554313 6
chr1 554313 554314 1
chr1 554315 554316 6
chr1 554316 554317 5
chr1 554317 554318 1
chr1 554318 554319 2
chr1 554319 554321 6
chr1 554321 554323 1
chr1 554323 554334 7
to
$ bedtools genomecov -ibam NA18152.bam -bg -strand + | head
chr1 554385 554392 4
chr1 554392 554408 5
chr1 554408 554430 6
chr1 554430 554451 7
chr1 554451 554455 8
chr1 554455 554490 9
chr1 554490 554495 10
chr1 554495 554496 9
chr1 554496 554574 10
chr1 554574 554579 11
-scale Scaling coverage by a constant factor.¶
The -scale option allows one to scale the coverage observed in an interval
file by a constant factor. Each coverage value is multiplied by this factor
before being reported. This can be useful for normalizing coverage by,
e.g., metrics such as reads per million (RPM).
Compare:
$ bedtools genomecov -ibam NA18152.bam -bg | head
chr1 554304 554309 5
chr1 554309 554313 6
chr1 554313 554314 1
chr1 554315 554316 6
chr1 554316 554317 5
chr1 554317 554318 1
chr1 554318 554319 2
chr1 554319 554321 6
chr1 554321 554323 1
chr1 554323 554334 7
to
$ bedtools genomecov -ibam NA18152.bam -bg -scale 10.0 | head
chr1 554304 554309 50
chr1 554309 554313 60
chr1 554313 554314 10
chr1 554315 554316 60
chr1 554316 554317 50
chr1 554317 554318 10
chr1 554318 554319 20
chr1 554319 554321 60
chr1 554321 554323 10
chr1 554323 554334 70
-split Reporting coverage with spliced alignments or blocked BED features¶
bedtools genomecov will, by default, screen for overlaps against the
entire span of a spliced/split BAM alignment or blocked BED12 feature. When
dealing with RNA-seq reads, for example, one typically wants to only screen
for overlaps for the portions of the reads that come from exons (and ignore the
interstitial intron sequence). The -split command allows for such
overlaps to be performed.
Coverage by fragment¶
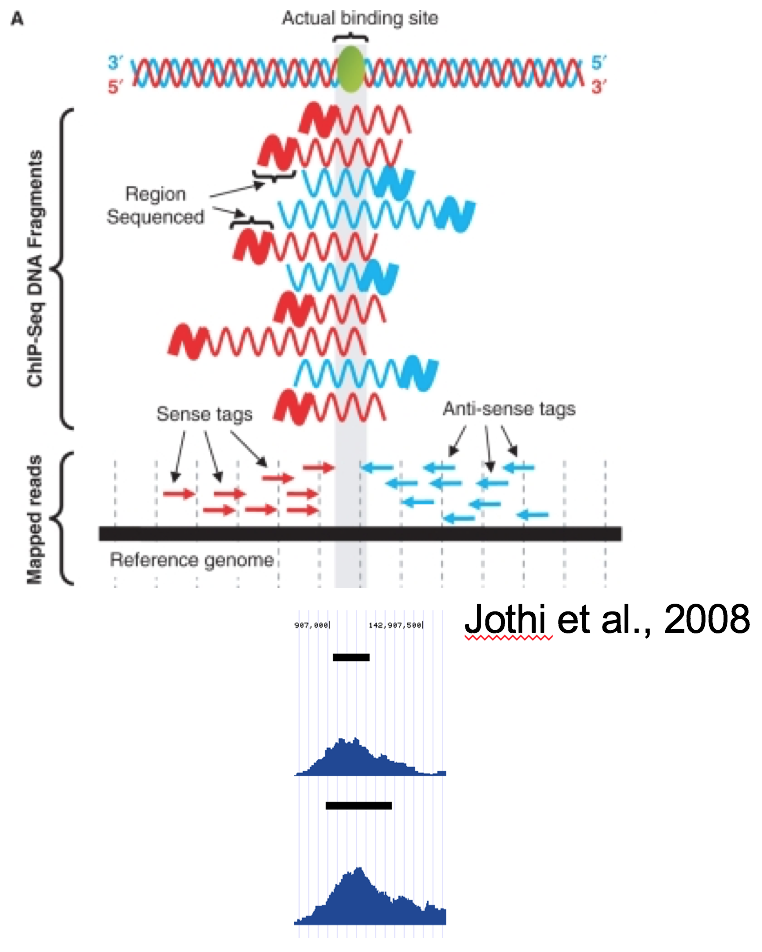
In ChiP-Seq the binding site is usually not at the coordinate where reads map, but in the middle of the fragment. For this reason we often try to estimate average fragment size for single-read experiment and extend the reads in the 5’-3’ direction up to the estimated fragment length. The coverage “by estimated fragments” or by actual pair-end fragments graph is expected to peak at the actual binding site.
-fs Forces to use provided fragment size.
-pc Calculates coverage for paired-end reads, coverage is calculated as the number of fragments covering each base pair
getfasta¶

bedtools getfasta extracts sequences from a FASTA file for each of the
intervals defined in a BED/GFF/VCF file.
Tip
1. The headers in the input FASTA file must exactly match the chromosome column in the BED file.
2. You can use the UNIX fold command to set the line width of the
FASTA output. For example, fold -w 60 will make each line of the FASTA
file have at most 60 nucleotides for easy viewing.
3. BED files containing a single region require a newline character at the end of the line, otherwise a blank output file is produced.
See also
Usage and option summary¶
Usage
$ bedtools getfasta [OPTIONS] -fi <input FASTA> -bed <BED/GFF/VCF>
(or):
$ getFastaFromBed [OPTIONS] -fi <input FASTA> -bed <BED/GFF/VCF>
| Option | Description |
|---|---|
| -fo | Specify an output file name. By default, output goes to stdout. |
| -name | Use the name field and coordinates for the FASTA header |
| -name+ | (deprecated) Use the name field and coordinates for the FASTA header |
| -nameOnly | Use the name field for the FASTA header |
| -tab | Report extract sequences in a tab-delimited format instead of in FASTA format. |
| -bedOut | Report extract sequences in a tab-delimited BED format instead of in FASTA format. |
| -s | Force strandedness. If the feature occupies the antisense strand, the sequence will be reverse complemented. Default: strand information is ignored. |
| -split | Given BED12 input, extract and concatenate the sequences from the BED “blocks” (e.g., exons) |
| -fullHeader | Use full fasta header. By default, only the word before the first space or tab is used. |
| -rna | The FASTA is RNA not DNA. Reverse complementation handled accordingly. |
Default behavior¶
bedtools getfasta will extract the sequence defined by the coordinates
in a BED interval and create a new FASTA entry in the output file for each
extracted sequence. By default, the FASTA header for each
extracted sequence will be formatted as follows: “<chrom>:<start>-<end>”.
$ cat test.fa
>chr1
AAAAAAAACCCCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
$ cat test.bed
chr1 5 10
$ bedtools getfasta -fi test.fa -bed test.bed
>chr1:5-10
AAACC
# optionally write to an output file
$ bedtools getfasta -fi test.fa -bed test.bed -fo test.fa.out
$ cat test.fa.out
>chr1:5-10
AAACC
-name Using the BED “name” column as a FASTA header.¶
Using the -name option, one can set the FASTA header for each extracted
sequence to be the “name” columns from the BED feature.
$ cat test.fa
>chr1
AAAAAAAACCCCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
$ cat test.bed
chr1 5 10 myseq
$ bedtools getfasta -fi test.fa -bed test.bed -name
>myseq
AAACC
-tab Creating a tab-delimited output file in lieu of FASTA output.¶
Using the -tab option, the -fo output file will be tab-delimited
instead of in FASTA format.
$ cat test.fa
>chr1
AAAAAAAACCCCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
$ cat test.bed
chr1 5 10 myseq
$ bedtools getfasta -fi test.fa -bed test.bed -name -tab
myseq AAACC
-bedOut Creating a tab-delimited BED file in lieu of FASTA output.¶
Using the -tab option, the -fo output file will be tab-delimited
instead of in FASTA format.
$ cat test.fa
>chr1
AAAAAAAACCCCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
$ cat test.bed
chr1 5 10 myseq
$ bedtools getfasta -fi test.fa -bed test.bed -tab
chr1 5 10 AAACC
-s Forcing the extracted sequence to reflect the requested strand¶
bedtools getfasta will extract the sequence in the orientation defined in
the strand column when the “-s” option is used.
$ cat test.fa
>chr1
AAAAAAAACCCCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
$ cat test.bed
chr1 20 25 forward 1 +
chr1 20 25 reverse 1 -
$ bedtools getfasta -fi test.fa -bed test.bed -s -name
>forward
CGCTA
>reverse
TAGCG
-split Extracting BED “blocks”.¶
One can optionally request that FASTA records be extracting and concatenating
each block in a BED12 record. For example, consider a BED12 record describing a
transcript. By default, getfasta will extract the sequence representing the
entire transcript (introns, exons, UTRs). Using the -split option, getfasta
will instead produce separate a FASTA record representing a transcript that
splices together each BED12 block (e.g., exons
and UTRs in the case of genes described with BED12).
$ cat genes.bed12
chr1 164404 173864 ENST00000466557.1 0 - 173864 173864 0 6 387,59,66,216,132,112, 0,1479,3695,4644,8152,9348,
chr1 235855 267253 ENST00000424587.1 0 - 267253 267253 0 4 2100,150,105,158, 0,2562,23161,31240,
chr1 317810 328455 ENST00000426316.1 0 + 328455 328455 0 2 323,145, 0,10500,
$ bedtools getfasta -fi chr1.fa -bed genes.bed12 -split -name
>ENST00000466557.1
gaggcgggaagatcacttgatatcaggagtcgaggcgggaagatcacttgacgtcaggagttcgagactggcccggccaacatggtgaaaccgcatctccactaaaaatacaaaaattagcctggtatggtggtgggcacctgtaatcccagtgacttgggaggctaaggcaggagaatttcttgaacccaggaggcagaggttgcagtgaccagcaaggttgcgccattgcaccccagcctgggcgataagagtgaaaactccatctcaaaaaaaaaaaaaaaaaaaaaaTTCCTTTGGGAAGGCCTTCTACATAAAAATCTTCAACATGAGACTGGAAAAAAGGGTATGGGATCATCACCGGACCTTTGGCTTTTACAGCTCGAGCTGACAAAGTTGATTTATCAAGTTGTAAATCTTCACCTGTTGAATTCATAAGTTCATGTCATATTTTCTTTCAGACAATTCTTCAGTTTGTTTACGTAGATCAGCGATACGATGATTCCATTTCTtcggatccttgtaagagcagagcaggtgatggagagggtgggaggtgtagtgacagaagcaggaaactccagtcattcgagacgggcagcacaagctgcggagtgcaggccacctctacggccaggaaacggattctcccgcagagcctcggaagctaccgaccctgctcccaccttgactcagtaggacttactgtagaattctggccttcagacCTGAGCCTGGCAGCTCTCTCCAACTTTGGAAGCCCAGGGGCATGGCCCCTGTCCACAGATGCACCTGGCATGAGGCGTGCCCAGAGGGACAGAGGCAGATGAGTttcgtctcctccactggattgtgagggcCAGAGTTGAACTCCCTCATTTTCCGTTCCCCAGCATTGGCAGGTTCTGGGACTGGTGGCTGTGGTGGCTCGTTGGTCTTTGTCTCTTAGAAGGTGGGGAATAATCATCATCT
>ENST00000424587.1
ccaggaagtgaaaatgacactttactgttttaatttgcatttctctgcttacaagtggattacacacattttcgtgtgctgttggctacttatTCATTCAGAAAACATACTAAGTGCTGGCTCTTTTTCATGTCCTTTATCAAGTTTGGATCATGTCATTTGCTATTTTCTTTCTGATGTAAACTCTCAAAGTCTGAAGTGTATTGTCTTTTCCTGACACATATGTTGTAAATAATTTTCTGGCTTACATTTTGACTTTTAATTTCATTCACGATGTTTTTAATGAATAATTTTAATTTTTATGAATGCAAGTTAAAATAATTCTTTCATTGTGGTCTCTGACATGTCATGCCAATAAGGGTCTTCTCCTCCAAGAGCACAGAAATATTTGCCAATACTGTCCTTAAAATCGGTCACAGTTTCATTTTTTATATATGCATTTTACTTCAATTGGGGCTTCATTTTACTGAATGCCCTATTTGAAGCAAGTTTCTCAGTTAATTCTTTTCTCAAAGGGCTAAGTATGGTAGATTGCAAACATAAGTGGCCACATAATGCTCTCACCTCctttgcctcctctcccaggaggagatagcgtccatctttccactccttaatctgggcttggccgtgtgacttgcactggccaatgggatattaacaagtctgatgtgcacagaggctgtagaatgtgcacgggggcttggtctctcttgctgccctggagaccagctgccCCACGAAGGAACCAGAGCCAACCTGCTGCTTCCTGGAGGAAGACAGTCCCTCTGTCCCTCTGTCTCTGCCAACCAGTTAACCTGCTGCTTCCTGGAGGGAGACAGTCCCTCAGTCCCTCTGTCTCTGCCAACCAGTTAACCTGCTGCTTCCTGGAGGAAGACAGTCACTCTGTCTCTGccaacccagttgaccgcagacatgcaggtctgctcaggtaagaccagcacagtccctgccctgtgagccaaaccaaatggtccagccacagaatcgtgagcaaataagtgatgcttaagtcactaagatttgggCAAAAGCTGAGCATTTATCCCAATCCCAATACTGTTTGTCCTTCTGTTTATCTGTCTGTCCTTCCCTGCTCATTTAAAATGCCCCCACTGCATCTAGTACATTTTTATAGGATCAGGGATCTGCTCTTGGATTAATGTTGTGTTCCCACCTCGAGGCAGCTTTGTAAGCTTCTGAGCACTTCCCAATTCCGGGTGACTTCAGGCACTGGGAGGCCTGTGCATCAGCTGCTGCTGTCTGTAGCTGACTTCCTTCACCCCTCTGCTGTCCTCAGCTCCTTCACCCCTGGGCCTCAGGAAATCAATGTCATGCTGACATCACTCTAGATCTAAAAGTTGGGTTCTTGgaccaggcgtggtggctcacacctgtaatcccagcactttgggaggccgaggcgggtggatcacaaggtcaggagatcaagacgattctggctaacacggtgaaaccccgtctctactaaaaatacaaaaaaattagccgggtgtggtggcaggtgcctgtagccccagctacttgggaggctgaggcaggagaatggcttgaacctgggaggtggagcttgcagtgagccaagatcacgccactgcactccagaatgggagagagagcgagactttctcaaaaaaaaaaaaaaaaCTTAGGTTCTTGGATGTTCGGGAAAGGGGGTTATTATCTAGGATCCTTGAAGCACCCCCAAGGGCATCTTCTCAAAGTTGGATGTGTGCATTTTCCTGAGAGGAAAGCTTTCCCACATTATACAGCTTCTGAAAGGGTTGCTTGACCCACAGATGTGAAGCTGAGGCTGAAGGAGACTGATGTGGTTTCTCCTCAGTTTCTCTGTGCAGCACCAGGTGGCAGCAGAGGTCAGCAAGGCAAACCCGAGCCCGGGGATGCGGAGTGGGGGCAGCTACGTCCTCTCTTGAGCTACAGCAGATTCACTCTGTTCTGTTTCATTGTTGTTTAGTTTGCGTTGTGTTTCTCCAACTTTGTGCCTCATCAGGAAAAGCTTTGGATCACAATTCCCAGtgctgaagaaaaggccaaactcttggttgtgttctttgattAGTgcctgtgacgcagcttcaggaggtcctgagaacgtgtgcacagtttagtcggcagaaacttagggaaatgtaagaccaccatcagcacataggagttctgcattggtttggtctgcattggtttggtCTTTTCCTGGATACAGGTCTTGATAGGTCTCTTGATGTCATTTCACTTCAGATTCTTCTTTAGAAAACTTGGACAATAGCATTTGCTGTCTTGTCCAAATTGTTACTTCAAGTTTGCTCTTAGCAAGTAATTGTTTCAGTATCTATATCAAAAATGGCTTAAGCCTGCAACATGTTTCTGAATGATTAACAAGGTGATAGTCAGTTCTTCATTGAATCCTGGATGCTTTATTTTTCTTAATAAGAGGAATTCATATGGATCAG
>ENST00000426316.1
AATGATCAAATTATGTTTCCCATGCATCAGGTGCAATGGGAAGCTCTTctggagagtgagagaagcttccagttaaggtgacattgaagccaagtcctgaaagatgaggaagagttgtatgagagtggggagggaagggggaggtggagggaTGGGGAATGGGCCGGGATGGGATAGCGCAAACTGCCCGGGAAGGGAAACCAGCACTGTACAGACCTGAACAACGAAGATGGCATATTTTGTTCAGGGAATGGTGAATTAAGTGTGGCAGGAATGCTTTGTAGACACAGTAATTTGCTTGTATGGAATTTTGCCTGAGAGACCTCATTCCTCACGTCGGCCATTCCAGGCCCCGTTTTTCCCTTCCGGCAGCCTCTTGGCCTCTAATTTGTTTATCTTTTGTGTATAAATCCCAAAATATTGAATTTTGGAATATTTCCACCATTATGTAAATATTTTGATAGGTAA
# use the UNIX fold command to wrap the FASTA sequence such that each line
# has at most 60 characters
$ bedtools getfasta -fi chr1.fa -bed genes.bed12 -split -name | \
fold -w 60
>ENST00000466557.1
gaggcgggaagatcacttgatatcaggagtcgaggcgggaagatcacttgacgtcaggag
ttcgagactggcccggccaacatggtgaaaccgcatctccactaaaaatacaaaaattag
cctggtatggtggtgggcacctgtaatcccagtgacttgggaggctaaggcaggagaatt
tcttgaacccaggaggcagaggttgcagtgaccagcaaggttgcgccattgcaccccagc
ctgggcgataagagtgaaaactccatctcaaaaaaaaaaaaaaaaaaaaaaTTCCTTTGG
GAAGGCCTTCTACATAAAAATCTTCAACATGAGACTGGAAAAAAGGGTATGGGATCATCA
CCGGACCTTTGGCTTTTACAGCTCGAGCTGACAAAGTTGATTTATCAAGTTGTAAATCTT
CACCTGTTGAATTCATAAGTTCATGTCATATTTTCTTTCAGACAATTCTTCAGTTTGTTT
ACGTAGATCAGCGATACGATGATTCCATTTCTtcggatccttgtaagagcagagcaggtg
atggagagggtgggaggtgtagtgacagaagcaggaaactccagtcattcgagacgggca
gcacaagctgcggagtgcaggccacctctacggccaggaaacggattctcccgcagagcc
tcggaagctaccgaccctgctcccaccttgactcagtaggacttactgtagaattctggc
cttcagacCTGAGCCTGGCAGCTCTCTCCAACTTTGGAAGCCCAGGGGCATGGCCCCTGT
CCACAGATGCACCTGGCATGAGGCGTGCCCAGAGGGACAGAGGCAGATGAGTttcgtctc
ctccactggattgtgagggcCAGAGTTGAACTCCCTCATTTTCCGTTCCCCAGCATTGGC
AGGTTCTGGGACTGGTGGCTGTGGTGGCTCGTTGGTCTTTGTCTCTTAGAAGGTGGGGAA
TAATCATCATCT
>ENST00000424587.1
ccaggaagtgaaaatgacactttactgttttaatttgcatttctctgcttacaagtggat
tacacacattttcgtgtgctgttggctacttatTCATTCAGAAAACATACTAAGTGCTGG
CTCTTTTTCATGTCCTTTATCAAGTTTGGATCATGTCATTTGCTATTTTCTTTCTGATGT
AAACTCTCAAAGTCTGAAGTGTATTGTCTTTTCCTGACACATATGTTGTAAATAATTTTC
TGGCTTACATTTTGACTTTTAATTTCATTCACGATGTTTTTAATGAATAATTTTAATTTT
TATGAATGCAAGTTAAAATAATTCTTTCATTGTGGTCTCTGACATGTCATGCCAATAAGG
GTCTTCTCCTCCAAGAGCACAGAAATATTTGCCAATACTGTCCTTAAAATCGGTCACAGT
TTCATTTTTTATATATGCATTTTACTTCAATTGGGGCTTCATTTTACTGAATGCCCTATT
TGAAGCAAGTTTCTCAGTTAATTCTTTTCTCAAAGGGCTAAGTATGGTAGATTGCAAACA
TAAGTGGCCACATAATGCTCTCACCTCctttgcctcctctcccaggaggagatagcgtcc
atctttccactccttaatctgggcttggccgtgtgacttgcactggccaatgggatatta
acaagtctgatgtgcacagaggctgtagaatgtgcacgggggcttggtctctcttgctgc
cctggagaccagctgccCCACGAAGGAACCAGAGCCAACCTGCTGCTTCCTGGAGGAAGA
CAGTCCCTCTGTCCCTCTGTCTCTGCCAACCAGTTAACCTGCTGCTTCCTGGAGGGAGAC
AGTCCCTCAGTCCCTCTGTCTCTGCCAACCAGTTAACCTGCTGCTTCCTGGAGGAAGACA
GTCACTCTGTCTCTGccaacccagttgaccgcagacatgcaggtctgctcaggtaagacc
agcacagtccctgccctgtgagccaaaccaaatggtccagccacagaatcgtgagcaaat
aagtgatgcttaagtcactaagatttgggCAAAAGCTGAGCATTTATCCCAATCCCAATA
CTGTTTGTCCTTCTGTTTATCTGTCTGTCCTTCCCTGCTCATTTAAAATGCCCCCACTGC
ATCTAGTACATTTTTATAGGATCAGGGATCTGCTCTTGGATTAATGTTGTGTTCCCACCT
CGAGGCAGCTTTGTAAGCTTCTGAGCACTTCCCAATTCCGGGTGACTTCAGGCACTGGGA
GGCCTGTGCATCAGCTGCTGCTGTCTGTAGCTGACTTCCTTCACCCCTCTGCTGTCCTCA
GCTCCTTCACCCCTGGGCCTCAGGAAATCAATGTCATGCTGACATCACTCTAGATCTAAA
AGTTGGGTTCTTGgaccaggcgtggtggctcacacctgtaatcccagcactttgggaggc
cgaggcgggtggatcacaaggtcaggagatcaagacgattctggctaacacggtgaaacc
ccgtctctactaaaaatacaaaaaaattagccgggtgtggtggcaggtgcctgtagcccc
agctacttgggaggctgaggcaggagaatggcttgaacctgggaggtggagcttgcagtg
agccaagatcacgccactgcactccagaatgggagagagagcgagactttctcaaaaaaa
aaaaaaaaaCTTAGGTTCTTGGATGTTCGGGAAAGGGGGTTATTATCTAGGATCCTTGAA
GCACCCCCAAGGGCATCTTCTCAAAGTTGGATGTGTGCATTTTCCTGAGAGGAAAGCTTT
CCCACATTATACAGCTTCTGAAAGGGTTGCTTGACCCACAGATGTGAAGCTGAGGCTGAA
GGAGACTGATGTGGTTTCTCCTCAGTTTCTCTGTGCAGCACCAGGTGGCAGCAGAGGTCA
GCAAGGCAAACCCGAGCCCGGGGATGCGGAGTGGGGGCAGCTACGTCCTCTCTTGAGCTA
CAGCAGATTCACTCTGTTCTGTTTCATTGTTGTTTAGTTTGCGTTGTGTTTCTCCAACTT
TGTGCCTCATCAGGAAAAGCTTTGGATCACAATTCCCAGtgctgaagaaaaggccaaact
cttggttgtgttctttgattAGTgcctgtgacgcagcttcaggaggtcctgagaacgtgt
gcacagtttagtcggcagaaacttagggaaatgtaagaccaccatcagcacataggagtt
ctgcattggtttggtctgcattggtttggtCTTTTCCTGGATACAGGTCTTGATAGGTCT
CTTGATGTCATTTCACTTCAGATTCTTCTTTAGAAAACTTGGACAATAGCATTTGCTGTC
TTGTCCAAATTGTTACTTCAAGTTTGCTCTTAGCAAGTAATTGTTTCAGTATCTATATCA
AAAATGGCTTAAGCCTGCAACATGTTTCTGAATGATTAACAAGGTGATAGTCAGTTCTTC
ATTGAATCCTGGATGCTTTATTTTTCTTAATAAGAGGAATTCATATGGATCAG
>ENST00000426316.1
AATGATCAAATTATGTTTCCCATGCATCAGGTGCAATGGGAAGCTCTTctggagagtgag
agaagcttccagttaaggtgacattgaagccaagtcctgaaagatgaggaagagttgtat
gagagtggggagggaagggggaggtggagggaTGGGGAATGGGCCGGGATGGGATAGCGC
AAACTGCCCGGGAAGGGAAACCAGCACTGTACAGACCTGAACAACGAAGATGGCATATTT
TGTTCAGGGAATGGTGAATTAAGTGTGGCAGGAATGCTTTGTAGACACAGTAATTTGCTT
GTATGGAATTTTGCCTGAGAGACCTCATTCCTCACGTCGGCCATTCCAGGCCCCGTTTTT
CCCTTCCGGCAGCCTCTTGGCCTCTAATTTGTTTATCTTTTGTGTATAAATCCCAAAATA
TTGAATTTTGGAATATTTCCACCATTATGTAAATATTTTGATAGGTAA
groupby¶
bedtools groupby is a useful tool that mimics the “group by” clause in database
systems. Given a file or stream that is sorted by the appropriate “grouping
columns” (-g), groupby will compute summary statistics on another
column (-c) in the file or stream. This will work with output from all
BEDTools as well as any other tab-delimited file or stream. As such, this is a
generally useful tool for all command-line analyses, not just genomics related
research.
Note
When using bedtools groupby, the input data must be ordered by the same
columns as specified with the -grp argument, which establish which
columns should be used to define a group of similar data. For example,
if -grp is 1,2,3, the data should be pre-grouped accordingly. When
bedtools groupby detects changes in the group columns it then summarizes
all lines with that group. For example, sort -k1,1 -k2,2 -k3,3 data.txt
| bedtools groupby -g 1,2,3 -c 4 -o mean.
Usage and option summary¶
Usage
bedtools groupby [OPTIONS] -i <input> -g <group columns> -c <op. column> -o <operation>
or:
groupBy [OPTIONS] -i <input> -g <group columns> -c <op. column> -o <operation>
| Option | Description |
|---|---|
| -i | The input file that should be grouped and summarized. Use “stdin” when using piped input. Note: if -i is omitted, input is assumed to come from standard input (stdin) |
| -g (-grp) | Specifies which column(s) (1-based) should be used to group the input. Columns may be comma-separated with each column must be explicitly listed. Or, ranges (e.g. 1-4) are also allowed. Default: 1,2,3 |
| -c (-opCol) | Specify the column (1-based) that should be summarized. Required. |
| -o (-op) | Specify the operation that should be applied to opCol. Valid operations:
sum - numeric only
count - numeric or text
count_distinct - numeric or text
min - numeric only
max - numeric only
mean - numeric only
median - numeric only
mode - numeric or text
antimode - numeric or text
stdev - numeric only
sstdev - numeric only
collapse (i.e., print a comma separated list) - numeric or text
distinct (i.e., print a comma separated list) - numeric or text
concat (i.e., print a comma separated list) - numeric or text
freqasc - print a comma separated list of values observed and the number of times they were observed.
Reported in ascending order of frequency*
freqdesc - print a comma separated list of values observed and the number of times they were observed.
Reported in descending order of frequency*
first - numeric or text
last - numeric or text
Default: sum
|
Default behavior.¶
Let’s imagine we have three incredibly interesting genetic variants that we are studying and we are interested in what annotated repeats these variants overlap.
$ cat variants.bed
chr21 9719758 9729320 variant1
chr21 9729310 9757478 variant2
chr21 9795588 9796685 variant3
$ bedtools intersect -a variants.bed -b repeats.bed -wa -wb > variantsToRepeats.bed
$ cat variantsToRepeats.bed
chr21 9719758 9729320 variant1 chr21 9719768 9721892 ALR/Alpha 1004 +
chr21 9719758 9729320 variant1 chr21 9721905 9725582 ALR/Alpha 1010 +
chr21 9719758 9729320 variant1 chr21 9725582 9725977 L1PA3 3288 +
chr21 9719758 9729320 variant1 chr21 9726021 9729309 ALR/Alpha 1051 +
chr21 9729310 9757478 variant2 chr21 9729320 9729809 L1PA3 3897 -
chr21 9729310 9757478 variant2 chr21 9729809 9730866 L1P1 8367 +
chr21 9729310 9757478 variant2 chr21 9730866 9734026 ALR/Alpha 1036 -
chr21 9729310 9757478 variant2 chr21 9734037 9757471 ALR/Alpha 1182 -
chr21 9795588 9796685 variant3 chr21 9795589 9795713 (GAATG)n 308 +
chr21 9795588 9796685 variant3 chr21 9795736 9795894 (GAATG)n 683 +
chr21 9795588 9796685 variant3 chr21 9795911 9796007 (GAATG)n 345 +
chr21 9795588 9796685 variant3 chr21 9796028 9796187 (GAATG)n 756 +
chr21 9795588 9796685 variant3 chr21 9796202 9796615 (GAATG)n 891 +
chr21 9795588 9796685 variant3 chr21 9796637 9796824 (GAATG)n 621 +
We can see that variant1 overlaps with 3 repeats, variant2 with 4 and variant3 with 6. We can use bedtools groupby to summarize the hits for each variant in several useful ways. The default behavior is to compute the sum of the opCol.
$ bedtools groupby -i variantsToRepeats.bed -g 1,2,3 -c 9
chr21 9719758 9729320 6353
chr21 9729310 9757478 14482
chr21 9795588 9796685 3604
Computing the min and max.¶
Now let’s find the min and max repeat score for each variant. We do this by “grouping” on the variant coordinate columns (i.e. cols. 1,2 and 3) and ask for the min and max of the repeat score column (i.e. col. 9).
$ bedtools groupby -i variantsToRepeats.bed -g 1,2,3 -c 9 -o min
chr21 9719758 9729320 1004
chr21 9729310 9757478 1036
chr21 9795588 9796685 308
We can also group on just the name column with similar effect.
$ bedtools groupby -i variantsToRepeats.bed -g 4 -c 9 -o min
variant1 1004
variant2 1036
variant3 308
What about the max score? Let’s keep the coordinates and the name of the variants so that we stay in BED format.
$ bedtools groupby -i variantsToRepeats.bed -grp 1-4 -c 9 -o max
chr21 9719758 9729320 variant1 3288
chr21 9729310 9757478 variant2 8367
chr21 9795588 9796685 variant3 891
Computing the mean and median.¶
Now let’s find the mean and median repeat score for each variant.
$ cat variantsToRepeats.bed | bedtools groupby -g 1-4 -c 9 -o mean
chr21 9719758 9729320 variant1 1588.25
chr21 9729310 9757478 variant2 3620.5
chr21 9795588 9796685 variant3 600.6667
$ bedtools groupby -i variantsToRepeats.bed -g 1-4 -c 9 -o median
chr21 9719758 9729320 variant1 1030.5
chr21 9729310 9757478 variant2 2539.5
chr21 9795588 9796685 variant3 652
Computing the mode and “antimode”.¶
Now let’s find the mode and antimode (i.e., the least frequent) repeat score for each variant (in this case they are identical).
$ bedtools groupby -i variantsToRepeats.bed -g 1-4 -c 9 -o mode
chr21 9719758 9729320 variant1 1004
chr21 9729310 9757478 variant2 1036
chr21 9795588 9796685 variant3 308
$ bedtools groupby -i variantsToRepeats.bed -g 1-4 -c 9 -o antimode
chr21 9719758 9729320 variant1 1004
chr21 9729310 9757478 variant2 1036
chr21 9795588 9796685 variant3 308
Computing the count of lines for a given group.¶
Figure:
$ bedtools groupby -i variantsToRepeats.bed -g 1-4 -c 9 -o count
chr21 9719758 9729320 variant1 4
chr21 9729310 9757478 variant2 4
chr21 9795588 9796685 variant3 6
Collapsing: listing all of the values in the opCol for a given group.¶
Now for something different. What if we wanted all of the names of the repeats listed on the same line as the variants? Use the collapse option. This “denormalizes” things. Now you have a list of all the repeats on a single line.
$ bedtools groupby -i variantsToRepeats.bed -grp 1-4 -c 9 -o collapse
chr21 9719758 9729320 variant1 ALR/Alpha,ALR/Alpha,L1PA3,ALR/Alpha,
chr21 9729310 9757478 variant2 L1PA3,L1P1,ALR/Alpha,ALR/Alpha,
chr21 9795588 9796685 variant3 (GAATG)n,(GAATG)n,(GAATG)n,(GAATG)n,(GAATG)n,(GAATG)n,
Computing frequencies: freqasc and freqdesc.¶
What if we want to report each distinct value along with its number of
occurrence (much like uniq -c)? The freqasc and freqdesc`` operations
handle this.
$ cat variantsToRepeats.bed | bedtools groupby -g 1 -c 8 -o freqdesc
chr21 (GAATG)n:6,ALR/Alpha:5,L1PA3:2,L1P1:1,
$ cat variantsToRepeats.bed | bedtools groupby -g 1 -c 8 -o freqasc
chr21 L1P1:1,L1PA3:2,ALR/Alpha:5,(GAATG)n:6,
igv¶
intersect¶
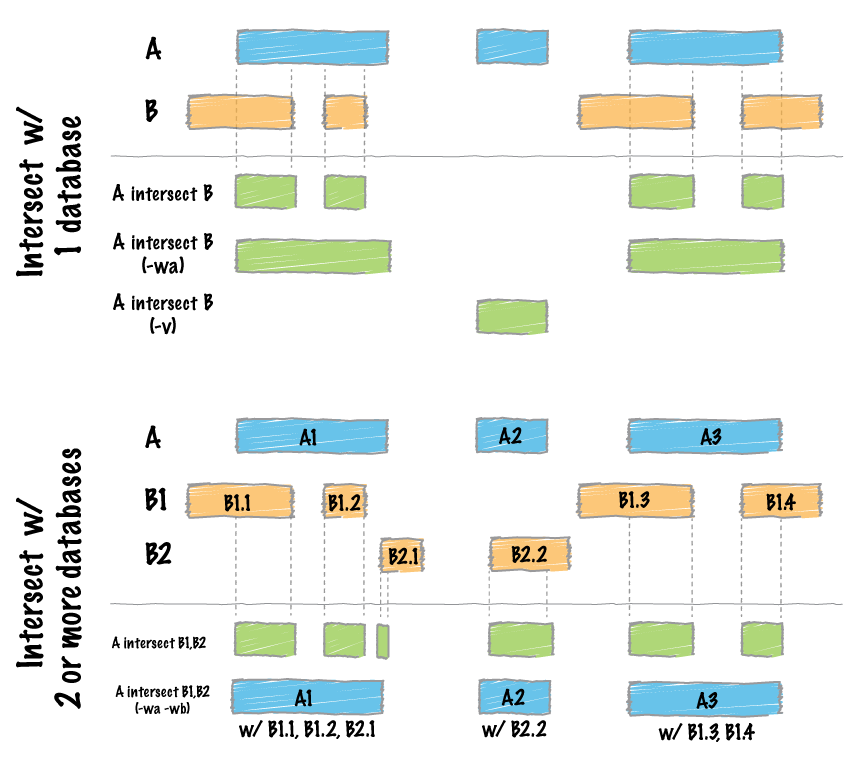
By far, the most common question asked of two sets of genomic features
is whether or not any of the features in the two sets “overlap”
with one another. This is known as feature intersection.
bedtools intersect allows one to screen for overlaps between
two sets of genomic features. Moreover, it allows one to have fine control
as to how the intersections are reported. bedtools intersect works
with both BED/GFF/VCF and BAM files as input.
Note
If you are trying to intersect very large files and are having trouble
with excessive memory usage, please presort your data by chromosome and
then by start position (e.g., sort -k1,1 -k2,2n in.bed > in.sorted.bed
for BED files) and then use the -sorted option. This invokes a
memory-efficient algorithm designed for large files. This algorithm has
been substantially improved in recent (>=2.18.0) releases.
Important
As of version 2.21.0, the intersect tool can accept multiple files for the -b option. This allows one to identify overlaps between a single query (-a) file and multiple database files (-b) at once!
Usage and option summary¶
Usage:
bedtools intersect [OPTIONS] -a <FILE> \
-b <FILE1, FILE2, ..., FILEN>
(or):
intersectBed [OPTIONS] -a <FILE> \
-b <FILE1, FILE2, ..., FILEN>
| Option | Description |
|---|---|
| -a | BAM/BED/GFF/VCF file “A”. Each feature in A is compared to B in search of overlaps. Use “stdin” if passing A with a UNIX pipe. |
| -b |
NEW!!!: -b may be followed with multiple databases and/or wildcard (*) character(s). |
| -abam | BAM file A. Each BAM alignment in A is compared to B in search of overlaps. Use “stdin” if passing A with a UNIX pipe: For example: samtools view -b <BAM> | bedtools intersect -abam stdin -b genes.bed. Note: no longer necessary after version 2.19.0 |
| -ubam | Write uncompressed BAM output. The default is write compressed BAM output. |
| -bed | When using BAM input (-abam), write output as BED. The default is to write output in BAM when using -abam. For example: bedtools intersect -abam reads.bam -b genes.bed -bed |
| -wa | Write the original entry in A for each overlap. |
| -wb | Write the original entry in B for each overlap. Useful for knowing what A overlaps. Restricted by -f and -r. |
| -loj | Perform a “left outer join”. That is, for each feature in A report each overlap with B. If no overlaps are found, report a NULL feature for B. |
| -wo | Write the original A and B entries plus the number of base pairs of overlap between the two features. Only A features with overlap are reported. Restricted by -f and -r. |
| -wao | Write the original A and B entries plus the number of base pairs of overlap between the two features. However, A features w/o overlap are also reported with a NULL B feature and overlap = 0. Restricted by -f and -r. |
| -u | Write original A entry once if any overlaps found in B. In other words, just report the fact at least one overlap was found in B. Restricted by -f and -r. |
| -c | For each entry in A, report the number of hits in B while restricting to -f. Reports 0 for A entries that have no overlap with B. Restricted -f, -F, -r, and -s. |
| -C | For each entry in A, separately report the number of overlaps with each B file on a distinct line. Reports 0 for A entries that have no overlap with B. Overlaps restricted by -f, -F, -r, and -s. |
| -v | Only report those entries in A that have no overlap in B. Restricted by -f and -r. |
| -f | Minimum overlap required as a fraction of A. Default is 1E-9 (i.e. 1bp). |
| -F | Minimum overlap required as a fraction of B. Default is 1E-9 (i.e., 1bp). |
| -r | Require that the fraction of overlap be reciprocal for A and B. In other words, if -f is 0.90 and -r is used, this requires that B overlap at least 90% of A and that A also overlaps at least 90% of B. |
| -e | Require that the minimum fraction be satisfied for A _OR_ B. In other words, if -e is used with -f 0.90 and -F 0.10 this requires that either 90% of A is covered OR 10% of B is covered. Without -e, both fractions would have to be satisfied. |
| -s | Force “strandedness”. That is, only report hits in B that overlap A on the same strand. By default, overlaps are reported without respect to strand. |
| -S | Require different strandedness. That is, only report hits in B that overlap A on the _opposite_ strand. By default, overlaps are reported without respect to strand. |
| -split | Treat “split” BAM (i.e., having an “N” CIGAR operation) or BED12 entries as distinct BED intervals. |
| -sorted | For very large B files, invoke a “sweeping” algorithm that requires position-sorted (e.g., sort -k1,1 -k2,2n for BED files) input. When using -sorted, memory usage remains low even for very large files. |
| -g | Specify a genome file the defines the expected chromosome order in the input files for use with the -sorted option. |
| -header | Print the header from the A file prior to results. |
| -names | When using multiple databases (-b), provide an alias for each that will appear instead of a fileId when also printing the DB record. |
| -filenames | When using multiple databases (-b), show each complete filename instead of a fileId when also printing the DB record. |
| -sortout | When using multiple databases (-b), sort the output DB hits for each record. |
| -nobuf | Disable buffered output. Using this option will cause each line of output to be printed as it is generated, rather than saved in a buffer. This will make printing large output files noticeably slower, but can be useful in conjunction with other software tools and scripts that need to process one line of bedtools output at a time. |
| -iobuf | Follow with desired integer size of read buffer. Optional suffixes K/M/G supported. Note: currently has no effect with compressed files. |
Default behavior¶
By default, if an overlap is found, bedtools intersect reports the shared interval between the two
overlapping features.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed
chr1 15 20
Intersecting against MULTIPLE -b files.¶
As of version 2.21.0, the intersect tool can detect overlaps between a single -a file and multiple -b files (instead of just one previously). One simply provides multiple -b files on the command line.
For example, consider the following query (-a) file and three distinct (-b) files:
$ cat query.bed
chr1 1 20
chr1 40 45
chr1 70 90
chr1 105 120
chr2 1 20
chr2 40 45
chr2 70 90
chr2 105 120
chr3 1 20
chr3 40 45
chr3 70 90
chr3 105 120
chr3 150 200
chr4 10 20
$ cat d1.bed
chr1 5 25
chr1 65 75
chr1 95 100
chr2 5 25
chr2 65 75
chr2 95 100
chr3 5 25
chr3 65 75
chr3 95 100
$ cat d2.bed
chr1 40 50
chr1 110 125
chr2 40 50
chr2 110 125
chr3 40 50
chr3 110 125
$ cat d3.bed
chr1 85 115
chr2 85 115
chr3 85 115
We can now compare query.bed to all three database files at once.:
$ bedtools intersect -a query.bed \
-b d1.bed d2.bed d3.bed
chr1 5 20
chr1 40 45
chr1 70 75
chr1 85 90
chr1 110 120
chr1 105 115
chr2 5 20
chr2 40 45
chr2 70 75
chr2 85 90
chr2 110 120
chr2 105 115
chr3 5 20
chr3 40 45
chr3 70 75
chr3 85 90
chr3 110 120
chr3 105 115
Clearly this is not completely informative because we cannot tell from which file each intersection came. However, if we use -wa and -wb, this becomes abundantly clear. When these options are used, the first column after the complete -a record lists the file number from which the overlap came. The number corresponds to the order in which the files were given on the command line.
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted
chr1 1 20 1 chr1 5 25
chr1 40 45 2 chr1 40 50
chr1 70 90 1 chr1 65 75
chr1 70 90 3 chr1 85 115
chr1 105 120 2 chr1 110 125
chr1 105 120 3 chr1 85 115
chr2 1 20 1 chr2 5 25
chr2 40 45 2 chr2 40 50
chr2 70 90 1 chr2 65 75
chr2 70 90 3 chr2 85 115
chr2 105 120 2 chr2 110 125
chr2 105 120 3 chr2 85 115
chr3 1 20 1 chr3 5 25
chr3 40 45 2 chr3 40 50
chr3 70 90 1 chr3 65 75
chr3 70 90 3 chr3 85 115
chr3 105 120 2 chr3 110 125
chr3 105 120 3 chr3 85 115
In many cases, it may be more useful to report an informative “label” for each file instead of a file number. One can do this with the -names option.
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-names d1 d2 d3 \
-sorted
chr1 1 20 d1 chr1 5 25
chr1 40 45 d2 chr1 40 50
chr1 70 90 d1 chr1 65 75
chr1 70 90 d3 chr1 85 115
chr1 105 120 d2 chr1 110 125
chr1 105 120 d3 chr1 85 115
chr2 1 20 d1 chr2 5 25
chr2 40 45 d2 chr2 40 50
chr2 70 90 d1 chr2 65 75
chr2 70 90 d3 chr2 85 115
chr2 105 120 d2 chr2 110 125
chr2 105 120 d3 chr2 85 115
chr3 1 20 d1 chr3 5 25
chr3 40 45 d2 chr3 40 50
chr3 70 90 d1 chr3 65 75
chr3 70 90 d3 chr3 85 115
chr3 105 120 d2 chr3 110 125
chr3 105 120 d3 chr3 85 115
Or perhaps it may be more useful to report the file name. One can do this with the -filenames option.
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted \
-filenames
chr1 1 20 d1.bed chr1 5 25
chr1 40 45 d2.bed chr1 40 50
chr1 70 90 d1.bed chr1 65 75
chr1 70 90 d3.bed chr1 85 115
chr1 105 120 d2.bed chr1 110 125
chr1 105 120 d3.bed chr1 85 115
chr2 1 20 d1.bed chr2 5 25
chr2 40 45 d2.bed chr2 40 50
chr2 70 90 d1.bed chr2 65 75
chr2 70 90 d3.bed chr2 85 115
chr2 105 120 d2.bed chr2 110 125
chr2 105 120 d3.bed chr2 85 115
chr3 1 20 d1.bed chr3 5 25
chr3 40 45 d2.bed chr3 40 50
chr3 70 90 d1.bed chr3 65 75
chr3 70 90 d3.bed chr3 85 115
chr3 105 120 d2.bed chr3 110 125
chr3 105 120 d3.bed chr3 85 115
Other options to intersect can be used as well. For example, let’s use -v to report those intervals in query.bed that do not overlap any of the intervals in the three database files:
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted \
-v
chr3 150 200
chr4 10 20
Or, let’s report only those intersections where 100% of the query record is overlapped by a database record:
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted \
-names d1 d2 d3
-f 1.0
chr1 40 45 d2 chr1 40 50
chr2 40 45 d2 chr2 40 50
chr3 40 45 d2 chr3 40 50
-wa Reporting the original A feature¶
Instead, one can force bedtools intersect to report the original “A” feature when an overlap is found. As
shown below, the entire “A” feature is reported, not just the portion that overlaps with the “B” feature.
For example:
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed -wa
chr1 10 20
-wb Reporting the original B feature¶
Similarly, one can force bedtools intersect to report the original “B” feature when an overlap is found. If
just -wb is used, the overlapping portion of A will be reported followed by the original “B”. If both -wa
and -wb are used, the originals of both “A” and “B” will be reported.
For example (-wb alone):
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed -wb
chr1 15 20 chr1 15 20
Now -wa and -wb:
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed -wa -wb
chr1 10 20 chr1 15 20
-loj Left outer join. Report features in A with and without overlaps¶
By default, bedtools intersect will only report features in A that
have an overlap in B. The -loj option will report every A feature
no matter what. When there is an overlap (or more than 1), it will report
A with its overlaps. Yet when there are no overlaps, an A feature will be
reported with a NULL B feature to indicate that there were no overlaps
For example (without -loj):
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed
chr1 10 20 chr1 15 20
Now with -loj:
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed -loj
chr1 10 20 chr1 15 20
chr1 30 40 . -1 -1
-wo Write the amount of overlap between intersecting features¶
The -wo option reports a column after each combination of intersecting
“A” and “B” features indicating the amount of overlap in bases pairs that
is observed between the two features.
Note
When an interval in A does not intersect an interval in B, it will not be
reported. If you would like to report such intervals with an overlap equal
to 0, see the -wao option.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ bedtools intersect -a A.bed -b B.bed -wo
chr1 10 20 chr1 15 20 5
chr1 10 20 chr1 18 25 2
-wao Write amounts of overlap for all features.¶
The -wao option extends upon the -wo option in that, unlike -wo,
it reports an overlap of 0 for features in A that do not have an intersection
in B.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ bedtools intersect -a A.bed -b B.bed -wao
chr1 10 20 chr1 15 20 5
chr1 10 20 chr1 18 25 2
chr1 30 40 . -1 -1 0
-u (unique) Reporting the mere presence of any overlapping features¶
Often you’d like to simply know a feature in “A” overlaps one or more
features in B without reporting each and every intersection. The -u
option will do exactly this: if an one or more overlaps exists, the
A feature is reported. Otherwise, nothing is reported.
For example, without -u:
$ cat A.bed
chr1 10 20
$ cat B.bed
chr1 15 20
chr1 17 22
$ bedtools intersect -a A.bed -b B.bed
chr1 15 20
chr1 17 20
Now with -u:
$ cat A.bed
chr1 10 20
$ cat B.bed
chr1 15 20
chr1 17 22
$ bedtools intersect -a A.bed -b B.bed -u
chr1 10 20
-c Reporting the number of overlapping features¶
The -c option reports a column after each “A” feature indicating the number (0 or more) of overlapping features found in “B”. Therefore, each feature in A is reported once.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ bedtools intersect -a A.bed -b B.bed -c
chr1 10 20 2
chr1 30 40 0
The -c option can also work with multiple -B files. In this case, the reported count will reflect the total number of intersections observed across _all_ -B files. It will not report a separate count for each database file. This can be achieved with the -C option.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ cat C.bed
chr1 16 21
chr1 19 26
$ bedtools intersect -a A.bed -b B.bed -c
chr1 10 20 4
chr1 30 40 0
-C Reporting the number of overlapping features for each database file¶
Unlike the the -c option, in the case of multiple -B files, the -C option will report a separate count for each database file.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ cat C.bed
chr1 16 21
chr1 19 26
$ bedtools intersect -a A.bed -b B.bed -C
chr1 10 20 1 2
chr1 10 20 2 2
chr1 30 40 1 0
chr1 30 40 1 0
If you would like to see more useful information than the file _number_ from which the counts came, one can use the -filenames or -names options.
$ bedtools intersect -a A.bed -b B.bed -C -filenames
chr1 10 20 A.bed 2
chr1 10 20 B.bed 2
chr1 30 40 A.bed 0
chr1 30 40 B.bed 0
$ bedtools intersect -a A.bed -b B.bed -C -names a b
chr1 10 20 a 2
chr1 10 20 b 2
chr1 30 40 a 0
chr1 30 40 b 0
-v Reporting the absence of any overlapping features¶
There will likely be cases where you’d like to know which “A” features
do not overlap with any of the “B” features. Perhaps you’d like to know
which SNPs don’t overlap with any gene annotations. The -v
(an homage to “grep -v”) option will only report those “A” features
that have no overlaps in “B”.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed -v
chr1 30 40
-f Requiring a minimal overlap fraction¶
By default, bedtools intersect will report an overlap between A and B so long as there is at least one base
pair is overlapping. Yet sometimes you may want to restrict reported overlaps between A and B to cases
where the feature in B overlaps at least X% (e.g. 50%) of the A feature. The -f option does exactly
this.
For example (note that the second B entry is not reported):
$ cat A.bed
chr1 100 200
$ cat B.bed
chr1 130 201
chr1 180 220
$ bedtools intersect -a A.bed -b B.bed -f 0.50 -wa -wb
chr1 100 200 chr1 130 201
-r, and -f Requiring reciprocal minimal overlap fraction¶
Similarly, you may want to require that a minimal fraction of both the A and the B features is overlapped. For example, if feature A is 1kb and feature B is 1Mb, you might not want to report the overlap as feature A can overlap at most 1% of feature B. If one set -f to say, 0.02, and one also enable the -r (reciprocal overlap fraction required), this overlap would not be reported.
For example (note that the second B entry is not reported):
$ cat A.bed
chr1 100 200
$ cat B.bed
chr1 130 201
chr1 130 200000
$ bedtools intersect -a A.bed -b B.bed -f 0.50 -r -wa -wb
chr1 100 200 chr1 130 201
-s Enforcing same strandedness¶
By default, bedtools intersect will report overlaps between features
even if the features are on opposite strands. However, if strand information
is present in both BED files and the “-s” option is used, overlaps will only
be reported when features are on the same strand.
For example (note that the first B entry is not reported):
$ cat A.bed
chr1 100 200 a1 100 +
$ cat B.bed
chr1 130 201 b1 100 -
chr1 132 203 b2 100 +
$ bedtools intersect -a A.bed -b B.bed -wa -wb -s
chr1 100 200 a1 100 + chr1 132 203 b2 100 +
-S Enforcing opposite “strandedness”¶
The -s option enforces that overlaps be on the same strand. In some
cases, you may want to enforce that overlaps be found on opposite strands.
In this, case use the -S option.
For example:
$ cat A.bed
chr1 100 200 a1 100 +
$ cat B.bed
chr1 130 201 b1 100 -
chr1 132 203 b2 100 +
$ bedtools intersect -a A.bed -b B.bed -wa -wb -S
chr1 100 200 a1 100 + chr1 130 201 b1 100 -
-abam Default behavior when using BAM input (deprecated since 2.18.0)¶
When comparing alignments in BAM format (-abam) to features in BED format (-b), bedtools intersect
will, by default, write the output in BAM format. That is, each alignment in the BAM file that meets
the user’s criteria will be written (to standard output) in BAM format. This serves as a mechanism to
create subsets of BAM alignments are of biological interest, etc. Note that only the mate in the BAM
alignment is compared to the BED file. Thus, if only one end of a paired-end sequence overlaps with a
feature in B, then that end will be written to the BAM output. By contrast, the other mate for the
pair will not be written. One should use pairToBed(Section 5.2) if one wants each BAM alignment
for a pair to be written to BAM output.
$ bedtools intersect -abam reads.unsorted.bam -b simreps.bed | \
samtools view - | \
head -3
BERTHA_0001:3:1:15:1362#0 99 chr4 9236904 0 50M = 9242033 5 1 7 9
AGACGTTAACTTTACACACCTCTGCCAAGGTCCTCATCCTTGTATTGAAG W c T U ] b \ g c e g X g f c b f c c b d d g g V Y P W W _
\c`dcdabdfW^a^gggfgd XT:A:R NM:i:0 SM:i:0 AM:i:0 X0:i:19 X1:i:2 XM:i:0 XO:i:0 XG:i:0 MD:Z:50
BERTHA _0001:3:1:16:994#0 83 chr6 114221672 37 25S6M1I11M7S =
114216196 -5493 G A A A G G C C A G A G T A T A G A A T A A A C A C A A C A A T G T C C A A G G T A C A C T G T T A
gffeaaddddggggggedgcgeggdegggggffcgggggggegdfggfgf XT:A:M NM:i:3 SM:i:37 AM:i:37 XM:i:2 X O : i :
1 XG:i:1 MD:Z:6A6T3
BERTHA _0001:3:1:16:594#0 147 chr8 43835330 0 50M =
43830893 -4487 CTTTGGGAGGGCTTTGTAGCCTATCTGGAAAAAGGAAATATCTTCCCATG U
\e^bgeTdg_Kgcg`ggeggg_gggggggggddgdggVg\gWdfgfgff XT:A:R NM:i:2 SM:i:0 AM:i:0 X0:i:10 X1:i:7 X M : i :
2 XO:i:0 XG:i:0 MD:Z:1A2T45
Note
As of version 2.18.0, it is no longer necessary to specify a BAM input file via -abam.
Bedtools now autodetects this when -a is used.
-ubam Default behavior when using BAM input¶
The -ubam option writes uncompressed BAM output to stdout. This is
useful for increasing the speed of pipelines that accept the output of
bedtools intersect as input, since the receiving tool does not need to
uncompress the data.
-bed Output BED format when using BAM input¶
When comparing alignments in BAM format (-abam) to features in BED format (-b), bedtools intersect
will optionally write the output in BED format. That is, each alignment in the BAM file is converted
to a 6 column BED feature and if overlaps are found (or not) based on the user’s criteria, the BAM
alignment will be reported in BED format. The BED “name” field is comprised of the RNAME field in
the BAM alignment. If mate information is available, the mate (e.g., “/1” or “/2”) field will be
appended to the name. The “score” field is the mapping quality score from the BAM alignment.
$ bedtools intersect -abam reads.unsorted.bam -b simreps.bed -bed | head -20
chr4 9236903 9236953 BERTHA_0001:3:1:15:1362#0/1 0 +
chr6 114221671 114221721 BERTHA_0001:3:1:16:994#0/1 37 -
chr8 43835329 43835379 BERTHA_0001:3:1:16:594#0/2 0 -
chr4 49110668 49110718 BERTHA_0001:3:1:31:487#0/1 23 +
chr19 27732052 27732102 BERTHA_0001:3:1:32:890#0/2 46 +
chr19 27732012 27732062 BERTHA_0001:3:1:45:1135#0/1 37 +
chr10 117494252 117494302 BERTHA_0001:3:1:68:627#0/1 37 -
chr19 27731966 27732016 BERTHA_0001:3:1:83:931#0/2 9 +
chr8 48660075 48660125 BERTHA_0001:3:1:86:608#0/2 37 -
chr9 34986400 34986450 BERTHA_0001:3:1:113:183#0/2 37 -
chr10 42372771 42372821 BERTHA_0001:3:1:128:1932#0/1 3 -
chr19 27731954 27732004 BERTHA_0001:3:1:130:1402#0/2 0 +
chr10 42357337 42357387 BERTHA_0001:3:1:137:868#0/2 9 +
chr1 159720631 159720681 BERTHA_0001:3:1:147:380#0/2 37 -
chrX 58230155 58230205 BERTHA_0001:3:1:151:656#0/2 37 -
chr5 142612746 142612796 BERTHA_0001:3:1:152:1893#0/1 37 -
chr9 71795659 71795709 BERTHA_0001:3:1:177:387#0/1 37 +
chr1 106240854 106240904 BERTHA_0001:3:1:194:928#0/1 37 -
chr4 74128456 74128506 BERTHA_0001:3:1:221:724#0/1 37 -
chr8 42606164 42606214 BERTHA_0001:3:1:244:962#0/1 37 +
-split Reporting overlaps with spliced alignments or blocked BED features¶
As described in section 1.3.19, bedtools intersect will, by default, screen for overlaps against the entire span of a spliced/split BAM alignment or blocked BED12 feature. When dealing with RNA-seq reads, for example, one typically wants to only screen for overlaps for the portions of the reads that come from exons (and ignore the interstitial intron sequence). The -split command allows for such overlaps to be performed.
For example, the diagram below illustrates the default behavior. The blue dots represent the “split/ spliced” portion of the alignment (i.e., CIGAR “N” operation). In this case, the two exon annotations are reported as overlapping with the “split” BAM alignment, but in addition, a third feature that overlaps the “split” portion of the alignment is also reported.
Chromosome ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Exons --------------- ----------
BED/BAM A ************.......................................****
BED File B ^^^^^^^^^^^^^^^ ^^^^^^^^ ^^^^^^^^^^
Result =============== ======== ==========
In contrast, when using the -split option, only the exon overlaps are reported.
Chromosome ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Exons --------------- ----------
BED/BAM A ************.......................................****
BED File B ^^^^^^^^^^^^^^^ ^^^^^^^^ ^^^^^^^^^^
Result =============== ==========
-sorted Invoke a memory-efficient algorithm for very large files.¶
The default algorithm for detecting overlaps loads the B file into an R-tree
structure in memory. While fast, it can consume substantial memory for large
files. For these reason, we provide an alternative, memory efficient algorithm
that depends upon inout files that have been sorted by chromosome and then by
start position. When both input files are position-sorted, the algorithm can
“sweep” through the data and detect overlaps on the fly in a manner much
like the way database systems join two tables. This option is invoked with the
-sorted option.
Note
By default, the -sorted option requires that the records are GROUPED
by chromosome and that within each chromosome group, the records are sorted by
chromosome position. One way to achieve this (for BED files for example) is use
the UNIX sort utility to sort both files by chromosome and then by position.
That is, sort -k1,1 -k2,2n in.bed > in.sorted.bed. However, since we merely
require that the chromsomes are grouped (that is, all records for a given chromosome
come in a single block in the file), sorting criteria other than the alphanumeric
criteria that is used by the sort utility are fine. For example, you could use
the “version sort” (-V) option in newer versions of GNU sort to make the chromosomes
come in this (chr1, chr2, chr3) order instead of this (chr1, chr10, chr11) order.
For example:
$ bedtools intersect -a big.sorted.bed -b huge.sorted.bed -sorted
-g Define an alternate chromosome sort order via a genome file.¶
As described above, the -sorted option expects that the input files are grouped
by chromosome. However, there arise cases where ones input
files are sorted by a different criteria and it is to computationally onerous
to resort the files alphanumerically. For example, the GATK expects that
BAM files are sorted in a very specific manner. The -g option allows
one to specify an exact ording that should be expected in the input (e.g.,
BAM, BED, etc.) files. All you need to do is re-order you genome file to
specify the order. Also, the use of a genome file to specify the expected
order allows the intersect tool to detect when two files are internally
grouped but each file actually follows a different order. This will cause
incorrect results and the -g file will alert you to such problems.
For example, an alphanumerically ordered genome file would look like the following:
$ cat hg19.genome
chr1 249250621
chr10 135534747
chr11 135006516
chr12 133851895
chr13 115169878
chr14 107349540
chr15 102531392
chr16 90354753
chr17 81195210
chr18 78077248
chr19 59128983
chr2 243199373
chr20 63025520
chr21 48129895
chr22 51304566
chr3 198022430
chr4 191154276
chr5 180915260
chr6 171115067
chr7 159138663
chr8 146364022
chr9 141213431
chrM 16571
chrX 155270560
chrY 59373566
However, if your input BAM or BED files are ordered such as chr1, chr2, chr3, etc.,
one need to simply reorder the genome file accordingly:
$ sort -k1,1V hg19.genome > hg19.versionsorted.genome
$ cat hg19.versionsorted.genome
chr1 249250621
chr2 243199373
chr3 198022430
chr4 191154276
chr5 180915260
chr6 171115067
chr7 159138663
chr8 146364022
chr9 141213431
chr10 135534747
chr11 135006516
chr12 133851895
chr13 115169878
chr14 107349540
chr15 102531392
chr16 90354753
chr17 81195210
chr18 78077248
chr19 59128983
chr20 63025520
chr21 48129895
chr22 51304566
chrM 16571
chrX 155270560
chrY 59373566
At this point, one can now use the -sorted option along with the genome file
in order to properly process the input files that abide by something other than an
alphanumeric sorting order.
$ bedtools intersect -a a.versionsorted.bam -b b.versionsorted.bed \
-sorted \
-g hg19.versionsorted.genome
Et voila.
-header Print the header for the A file before reporting results.¶
By default, if your A file has a header, it is ignored when reporting results. This option will instead tell bedtools to first print the header for the A file prior to reporting results.
jaccard¶
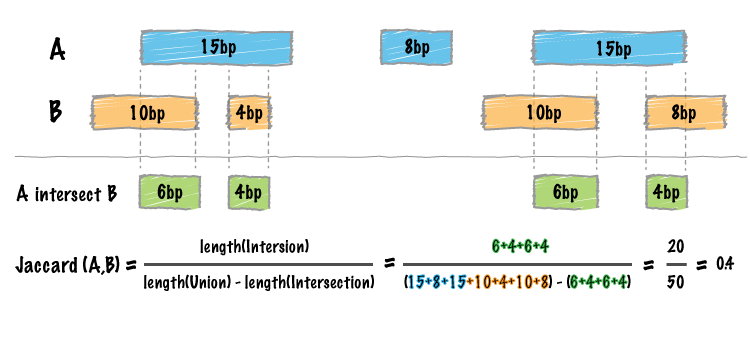
Whereas the bedtools intersect tool enumerates each an every intersection between two sets of genomic
intervals, one often needs a single statistic reflecting the similarity of the two sets based on the
intersections between them. The Jaccard statistic is used in set theory to represent the ratio of the
intersection of two sets to the union of the two sets. Similarly, Favorov et al [1] reported the use
of the Jaccard statistic for genome intervals: specifically, it measures the ratio of the number of
intersecting base pairs between two sets to the number of base pairs in the union of the two sets.
The bedtools jaccard tool implements this statistic, yet modifies the statistic such that the length of the
intersection is subtracted from the length of the union. As a result, the final statistic ranges from 0.0
to 1.0, where 0.0 represents no overlap and 1.0 represent complete overlap.
[1] Exploring Massive, Genome Scale Datasets with the GenometriCorr Package.
Favorov A, Mularoni L, Cope LM, Medvedeva Y, Mironov AA, et al. (2012)
PLoS Comput Biol 8(5): e1002529. doi:10.1371/journal.pcbi.1002529
Note
The jaccard tool requires that your data is pre-sorted by chromosome and
then by start position (e.g., sort -k1,1 -k2,2n in.bed > in.sorted.bed
for BED files).
Usage and option summary¶
Usage:
bedtools jaccard [OPTIONS] -a <BED/GFF/VCF> -b <BED/GFF/VCF>
| Option | Description |
|---|---|
| -a | BED/GFF/VCF file A. Each feature in A is compared to B in search of overlaps. Use “stdin” if passing A with a UNIX pipe. |
| -b | BED/GFF/VCF file B. Use “stdin” if passing B with a UNIX pipe. |
| -f | Minimum overlap required as a fraction of A. Default is 1E-9 (i.e. 1bp). |
| -F | Minimum overlap required as a fraction of B. Default is 1E-9 (i.e., 1bp). |
| -r | Require that the fraction of overlap be reciprocal for A and B. In other words, if -f is 0.90 and -r is used, this requires that B overlap at least 90% of A and that A also overlaps at least 90% of B. |
| -e | Require that the minimum fraction be satisfied for A _OR_ B. In other words, if -e is used with -f 0.90 and -F 0.10 this requires that either 90% of A is covered OR 10% of B is covered. Without -e, both fractions would have to be satisfied.**-s** Force “strandedness”. That is, only report hits in B that overlap A on the same strand. By default, overlaps are reported without respect to strand. |
| -S | Require different strandedness. That is, only report hits in B that overlap A on the _opposite_ strand. By default, overlaps are reported without respect to strand. |
| -split | Treat “split” BAM (i.e., having an “N” CIGAR operation) or BED12 entries as distinct BED intervals. |
Default behavior¶
By default, bedtools jaccard reports the length of the intersection, the length of the union (minus the intersection),
the final Jaccard statistic reflecting the similarity of the two sets, as well as the number of intersections.
$ cat a.bed
chr1 10 20
chr1 30 40
$ cat b.bed
chr1 15 20
$ bedtools jaccard -a a.bed -b b.bed
intersection union jaccard n_intersections
5 20 0.25 1
Controlling which intersections are included¶
One can also control which intersections are included in the statistic by requiring a certain fraction of overlap
with respect to the features in A (via the -f parameter) or also by requiring that the fraction of overlap is
reciprocal (-r) in A and B.
$ cat a.bed
chr1 10 20
chr1 30 40
$ cat b.bed
chr1 15 20
Require 10% overlap with respect to the intervals in A:
$ bedtools jaccard -a a.bed -b b.bed -f 0.1
intersection union jaccard n_intersections
5 20 0.25 1
Require 60% overlap with respect to the intervals in A:
$ bedtools jaccard -a a.bed -b b.bed -f 0.6
intersection union jaccard n_intersections
0 25 0.25 0
links¶
Creates an HTML file with links to an instance of the UCSC Genome Browser for all features / intervals in a file. This is useful for cases when one wants to manually inspect through a large set of annotations or features.
Usage and option summary¶
Usage:
linksBed [OPTIONS] -i <BED/GFF/VCF> > <HTML file>
| Option | Description |
|---|---|
| -base | The “basename” for the UCSC browser. Default: http://genome.ucsc.edu |
| -org | The organism (e.g. mouse, human). Default: human |
| -db | The genome build. Default: hg18 |
Default behavior¶
By default, linksBed creates links to the public UCSC Genome Browser.
For example:
head genes.bed
chr21 9928613 10012791 uc002yip.1 0 -
chr21 9928613 10012791 uc002yiq.1 0 -
chr21 9928613 10012791 uc002yir.1 0 -
chr21 9928613 10012791 uc010gkv.1 0 -
chr21 9928613 10061300 uc002yis.1 0 -
chr21 10042683 10120796 uc002yit.1 0 -
chr21 10042683 10120808 uc002yiu.1 0 -
chr21 10079666 10120808 uc002yiv.1 0 -
chr21 10080031 10081687 uc002yiw.1 0 -
chr21 10081660 10120796 uc002yix.2 0 -
linksBed -i genes.bed > genes.html
When genes.html is opened in a web browser, one should see something like the following, where each link on the page is built from the features in genes.bed:
Creating HTML links to a local UCSC Browser installation¶
Optionally, linksBed will create links to a local copy of the UCSC Genome Browser.
For example:
head -3 genes.bed
chr21 9928613 10012791 uc002yip.1 0 -
chr21 9928613 10012791 uc002yiq.1 0 -
linksBed -i genes.bed -base http://mirror.uni.edu > genes.html
One can point the links to the appropriate organism and genome build as well:
head -3 genes.bed
chr21 9928613 10012791 uc002yip.1 0 -
chr21 9928613 10012791 uc002yiq.1 0 -
linksBed -i genes.bed -base http://mirror.uni.edu -org mouse -db mm9 > genes.html
makewindows¶
map¶
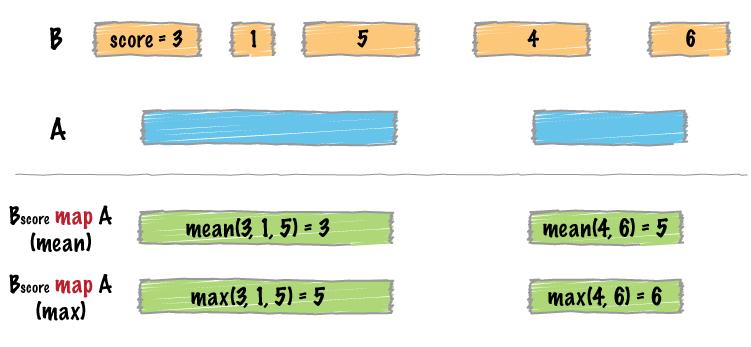
bedtools map allows one to map overlapping features in a B file onto
features in an A file and apply statistics and/or summary operations on those
features.
For example, one could use bedtools map to compute the average
score of BEDGRAPH records that overlap genes. Since the fourth column in
BEDGRAPH is the score, the following command illustrates how this would be done:
$ bedtools map -a genes.bed -b peaks.bedgraph -c 4 -o mean
Another example is discussed in this Biostars post.
Note
bedtools map requires each input file to be sorted by genome coordinate.
For BED files, this can be done with sort -k1,1 -k2,2n. Other sorting criteria are allowed if a genome file (-g) is provides that specifies the expected chromosome order.
Note
The map tool is substantially faster in versions 2.19.0 and later. The plot below demonstrates the increased speed when, for example, counting the number of exome alignments that align to each exon. The bedtools times are compared to the bedops bedmap utility as a point of reference.
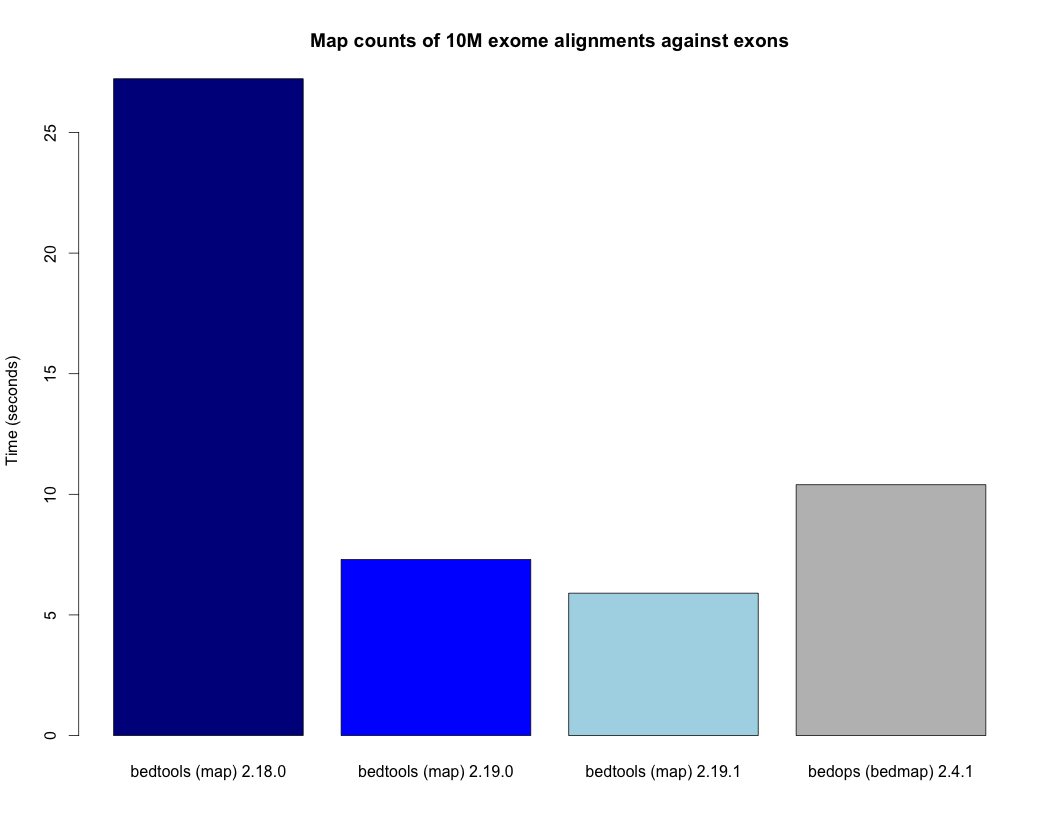
Usage and option summary¶
Usage:
bedtools map [OPTIONS] -a <bed/gff/vcf> -b <bed/gff/vcf>
(or):
mapBed [OPTIONS] -a <bed/gff/vcf> -b <bed/gff/vcf>
| Option | Description |
|---|---|
| -c | Specify the column from the B file to map onto intervals in A.
Default: 5 |
| -o | Specify the operation that should be applied to Valid operations:
sum - numeric only
count - numeric or text
count_distinct - numeric or text
min - numeric only
max - numeric only
absmin - numeric only
absmax - numeric only
mean - numeric only
median - numeric only
antimode - numeric or text
collapse (i.e., print a comma separated list) - numeric or text
distinct (i.e., print a comma separated list) - numeric or text
concat (i.e., print a comma separated list) - numeric or text
|
| -f | Minimum overlap required as a fraction of A. Default is 1E-9 (i.e. 1bp). |
| -F | Minimum overlap required as a fraction of B. Default is 1E-9 (i.e., 1bp). |
| -r | Require that the fraction of overlap be reciprocal for A and B. In other words, if -f is 0.90 and -r is used, this requires that B overlap at least 90% of A and that A also overlaps at least 90% of B. |
| -e | Require that the minimum fraction be satisfied for A _OR_ B. In other words, if -e is used with -f 0.90 and -F 0.10 this requires that either 90% of A is covered OR 10% of B is covered. Without -e, both fractions would have to be satisfied. |
| -s | Force “strandedness”. That is, only report hits in B that overlap A on the same strand. By default, overlaps are reported without respect to strand. |
| -S | Require different strandedness. That is, only report hits in B that overlap A on the _opposite_ strand. By default, overlaps are reported without respect to strand. |
| -null | The value to print if no overlaps are found for an A interval.
Default: "." |
| -header | Print the header from the A file prior to results. |
| -split | Treat “split” BAM (i.e., having an “N” CIGAR operation) or BED12 entries as distinct BED intervals. When using -sorted, memory usage remains low even for very large files. |
| -g | Specify a genome file the defines the expected chromosome order in the input files. |
Default behavior - compute the sum of the score column for all overlaps.¶
By default, map computes the sum of the 5th column (the score field for
BED format) for all intervals in B that overlap each interval in A.
Tip
Records in A that have no overlap will, by default, return . for the
computed value from B. This can be changed with the -null option.
$ cat a.bed
chr1 10 20 a1 1 +
chr1 50 60 a2 2 -
chr1 80 90 a3 3 -
$ cat b.bed
chr1 12 14 b1 2 +
chr1 13 15 b2 5 -
chr1 16 18 b3 5 +
chr1 82 85 b4 2 -
chr1 85 87 b5 3 +
$ bedtools map -a a.bed -b b.bed
chr1 10 20 a1 1 + 12
chr1 50 60 a2 2 - .
chr1 80 90 a3 3 - 5
mean Compute the mean of a column from overlapping intervals¶
$ cat a.bed
chr1 10 20 a1 1 +
chr1 50 60 a2 2 -
chr1 80 90 a3 3 -
$ cat b.bed
chr1 12 14 b1 2 +
chr1 13 15 b2 5 -
chr1 16 18 b3 5 +
chr1 82 85 b4 2 -
chr1 85 87 b5 3 +
$ bedtools map -a a.bed -b b.bed -c 5 -o mean
chr1 10 20 a1 1 + 4
chr1 50 60 a2 2 - .
chr1 80 90 a3 3 - 2.5
collapse List each value of a column from overlapping intervals¶
$ bedtools map -a a.bed -b b.bed -c 5 -o collapse
chr1 10 20 a1 1 + 2,5,5
chr1 50 60 a2 2 - .
chr1 80 90 a3 3 - 2,3
distinct List each unique value of a column from overlapping intervals¶
$ bedtools map -a a.bed -b b.bed -c 5 -o distinct
chr1 10 20 a1 1 + 2,5
chr1 50 60 a2 2 - .
chr1 80 90 a3 3 - 2,3
-s Only include intervals that overlap on the same strand.¶
$ bedtools map -a a.bed -b b.bed -c 5 -o collapse -s
chr1 10 20 a1 1 + 2,5
chr1 50 60 a2 2 - .
chr1 80 90 a3 3 - 2
-S Only include intervals that overlap on the opposite strand.¶
$ bedtools map -a a.bed -b b.bed -c 5 -o collapse -S
chr1 10 20 a1 1 + 5
chr1 50 60 a2 2 - .
chr1 80 90 a3 3 - 3
Multiple operations and columns at the same time.¶
As of version 2.19.1, multiple columns and operations are allowed at the same time in a single run. This greatly expedites analyses by preventing one from having to process the same file over an over for each column/operation.
$ bedtools map -a a.bed -b b.bed -c 5,5,5,5 -o min,max,median,collapse
Or, apply the same function to multiple columns:
$ bedtools map -a a.bed -b b.bed -c 3,4,5,6 -o mean
maskfasta¶
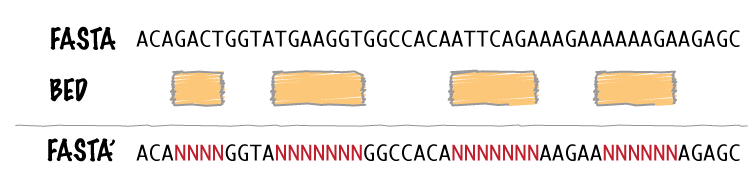
bedtools maskfasta masks sequences in a FASTA file based on intervals defined in a feature file. The
headers in the input FASTA file must exactly match the chromosome column in the feature file. This
may be useful for creating your own masked genome file based on custom annotations or for masking all
but your target regions when aligning sequence data from a targeted capture experiment.
Usage and option summary¶
Usage
$ bedtools maskfasta [OPTIONS] -fi <input FASTA> -bed <BED/GFF/VCF> -fo <output FASTA>
(or):
$ maskFastaFromBed [OPTIONS] -fi <input FASTA> -bed <BED/GFF/VCF> -fo <output FASTA>
Note
The input (-fi) and output (-fo) FASTA files must be different.
See also
| Option | Description |
|---|---|
| -soft | Soft-mask (that is, convert to lower-case bases) the FASTA sequence. By default, hard-masking (that is, conversion to Ns) is performed. |
| -mc | Replace masking character. That is, instead of masking with Ns, use another character. |
Default behavior¶
bedtools maskfasta will mask a FASTA file based on the intervals in a BED file. The newly masked FASTA file is written to the output FASTA file.
$ cat test.fa
>chr1
AAAAAAAACCCCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
$ cat test.bed
chr1 5 10
$ bedtools maskfasta -fi test.fa -bed test.bed -fo test.fa.out
$ cat test.fa.out
>chr1
AAAAANNNNNCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
-soft Soft-masking the FASTA file.¶
Using the -soft option, one can optionally “soft-mask” the FASTA file.
$ cat test.fa
>chr1
AAAAAAAACCCCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
$ cat test.bed
chr1 5 10
$ bedtools maskfasta -fi test.fa -bed test.bed -fo test.fa.out -soft
$ cat test.fa.out
>chr1
AAAAAaaaccCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
-mc Specify a masking character.¶
Using the -mc option, one can optionally choose a masking character to each base that will be masked by the BED file.
$ cat test.fa
>chr1
AAAAAAAACCCCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
$ cat test.bed
chr1 5 10
$ bedtools maskfasta -fi test.fa -bed test.bed -fo test.fa.out -mc X
$ cat test.fa.out
>chr1
AAAAAXXXXXCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
merge¶
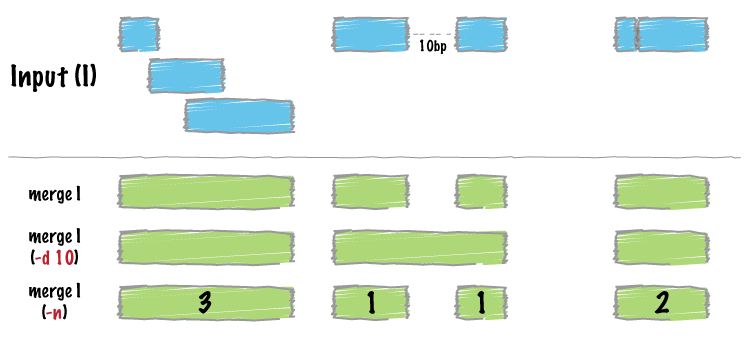
bedtools merge combines overlapping or “book-ended” features in an interval
file into a single feature which spans all of the combined features.
Note
bedtools merge requires that you presort your data by chromosome and
then by start position (e.g., sort -k1,1 -k2,2n in.bed > in.sorted.bed
for BED files).
See also
Usage and option summary¶
Usage:
bedtools merge [OPTIONS] -i <BED/GFF/VCF/BAM>
(or):
mergeBed [OPTIONS] -i <BED/GFF/VCF/BAM>
| Option | Description |
|---|---|
| -s | Force strandedness. That is, only merge features that are the same strand. By default, this is disabled. |
| -S | Force merge for one specific strand only. Follow with + or - to force merge from only the forward or reverse strand, respectively. By default, merging is done without respect to strand. |
| -d | Maximum distance between features allowed for features to be merged. Default is 0. That is, overlapping and/or book-ended features are merged. |
| -c | Specify columns from the input file to operate upon (see -o option, below). Multiple columns can be specified in a comma-delimited list. |
| -o | Specify the operation that should be applied to
-c.Valid operations:
sum, min, max, absmin, absmax,
mean, median,
collapse (i.e., print a delimited list (duplicates allowed)),
distinct (i.e., print a delimited list (NO duplicates allowed)),
count
count_distinct (i.e., a count of the unique values in the column),
Default: sum
Multiple operations can be specified in a comma-delimited list.
If there is only column, but multiple operations, all operations will be
applied on that column. Likewise, if there is only one operation, but
multiple columns, that operation will be applied to all columns.
Otherwise, the number of columns must match the the number of operations,
and will be applied in respective order.
E.g.,
-c 5,4,6 -o sum,mean,count will give the sum of column 5,the mean of column 4, and the count of column 6.
The order of output columns will match the ordering given in the command.
|
| -header | Print the header from the A file prior to results.
|
| -delim | Specify a custom delimiter for the -nms and -scores concat options
Example:
-delim "|"Default: ";" |
Default behavior¶
By default, bedtools merge combines overlapping (by at least 1 bp) and/or
bookended intervals into a single, “flattened” or “merged” interval.
$ cat A.bed
chr1 100 200
chr1 180 250
chr1 250 500
chr1 501 1000
$ bedtools merge -i A.bed
chr1 100 500
chr1 501 1000
-s Enforcing “strandedness”¶
The -s option will only merge intervals that are overlapping/bookended
and are on the same strand.
$ cat A.bed
chr1 100 200 a1 1 +
chr1 180 250 a2 2 +
chr1 250 500 a3 3 -
chr1 501 1000 a4 4 +
$ bedtools merge -i A.bed -s
chr1 100 250
chr1 501 1000
chr1 250 500
To also report the strand, you could use the -c and -o operators (see below for more details):
$ bedtools merge -i A.bed -s -c 6 -o distinct
chr1 100 250 +
chr1 501 1000 +
-S Reporting merged intervals on a specific strand.¶
The -S option will only merge intervals for a specific strand. For example,
to only report merged intervals on the “+” strand:
$ cat A.bed
chr1 100 200 a1 1 +
chr1 180 250 a2 2 +
chr1 250 500 a3 3 -
chr1 501 1000 a4 4 +
$ bedtools merge -i A.bed -S +
chr1 100 250
chr1 501 1000
To also report the strand, you could use the -c and -o operators (see below for more details):
$ bedtools merge -i A.bed -S + -c 6 -o distinct
chr1 100 250 +
chr1 501 1000 +
-d Controlling how close two features must be in order to merge¶
By default, only overlapping or book-ended features are combined into a new
feature. However, one can force merge to combine more distant features
with the -d option. For example, were one to set -d to 1000, any
features that overlap or are within 1000 base pairs of one another will be
combined.
$ cat A.bed
chr1 100 200
chr1 501 1000
$ bedtools merge -i A.bed
chr1 100 200
chr1 501 1000
$ bedtools merge -i A.bed -d 1000
chr1 100 200 1000
-c and -o Applying operations to columns from merged intervals.¶
When merging intervals, we often want to summarize or keep track of the
values observed in specific columns (e.g., the feature name or score) from
the original, unmerged intervals. When used together, the -c and -o
options allow one to select specific columns (-c) and apply operation
(-o) to each column. The result will be appended to the default, merged
interval output. For example, one could use the following to report the
count of intervals that we merged in each resulting interval (this replaces
the -n option that existed prior to version 2.20.0).
$ cat A.bed
chr1 100 200
chr1 180 250
chr1 250 500
chr1 501 1000
$ bedtools merge -i A.bed -c 1 -o count
chr1 100 500 3
chr1 501 1000 1
We could also use these options to report the mean of the score (#5) field:
$ cat A.bed
chr1 100 200 a1 1 +
chr1 180 250 a2 2 +
chr1 250 500 a3 3 -
chr1 501 1000 a4 4 +
$ bedtools merge -i A.bed -c 5 -o mean
chr1 100 500 2
chr1 501 1000 4
Let’s get fancy and report the mean, min, and max of the score column:
$ bedtools merge -i A.bed -c 5 -o mean,min,max
chr1 100 500 2 1 3
chr1 501 1000 4 4 4
Let’s also report a comma-separated list of the strands:
$ bedtools merge -i A.bed -c 5,5,5,6 -o mean,min,max,collapse
chr1 100 500 2 1 3 +,+,-
chr1 501 1000 4 4 4 +
Hopefully this provides a clear picture of what can be done.
-n Reporting the number of features that were merged¶
Deprecated since version 2.20.0.
See the -c and -o operators.
-nms Reporting the names of the features that were merged¶
Deprecated since version 2.20.0.
See the -c and -o operators.
-scores Reporting the scores of the features that were merged¶
Deprecated since version 2.20.0.
See the -c and -o operators.
-delim Change the delimiter for -c and -o¶
One can override the use of a comma as the delimiter for the -c and
-o collapse|distinct options via the use of the -delim option.
$ cat A.bed
chr1 100 200 A1
chr1 150 300 A2
chr1 250 500 A3
Compare:
$ bedtools merge -i A.bed -c 4 -o collapse
chr1 100 500 A1,A2,A3
to:
$ bedtools merge -i A.bed -c 4 -o collapse -delim "|"
chr1 100 500 A1|A2|A3
multicov¶
bedtools multicov, reports the count of alignments from multiple
position-sorted and indexed BAM files that overlap intervals in a BED file.
Specifically, for each BED interval provided, it reports a separate count of
overlapping alignments from each BAM file.
Note
bedtools multicov depends upon index BAM files in order to count the
number of overlaps in each BAM file. As such, each BAM file should be
position sorted (samtool sort aln.bam aln.sort) and
indexed (samtools index aln.sort.bam) with either samtools or bamtools.
Usage and option summary¶
Usage:
bedtools multicov [OPTIONS] -bams BAM1 BAM2 BAM3 ... BAMn -bed <BED/GFF/VCF>
(or):
multiBamCov [OPTIONS] -bams BAM1 BAM2 BAM3 ... BAMn -bed <BED/GFF/VCF>
| Option | Description |
|---|---|
| -split | Treat “split” BAM or BED12 entries as distinct BED intervals. |
| -s | Require same strandedness. That is, only report hits in B that overlap A on the _same_ strand. By default, overlaps are reported without respect to strand. |
| -S | Require different strandedness. That is, only report hits in B that overlap A on the _opposite_ strand. By default, overlaps are reported without respect to strand. |
| -f | Minimum overlap required as a fraction of each -bed record. Default is 1E-9 (i.e., 1bp). |
| -r | Require that the fraction overlap is reciprocal for -bed and B. In other words, if -f is 0.90 and -r is used, this requires that B overlap 90% of the -bed record and the -bed record _also_ overlaps 90% of B. |
| -q | Minimum mapping quality (MAPQ) allowed. Default is 0. |
| -D | Include duplicate reads. Default counts non-duplicates only |
| -F | Include failed-QC reads. Default counts pass-QC reads only |
| -p | Only count proper pairs. Default counts all alignments with
MAPQ > -q argument, regardless of the BAM FLAG field. |
Default behavior.¶
By default, multicov will report the count of alignments in each input
BAM file that overlap.
$ cat ivls-of-interest.bed
chr1 0 10000 ivl1
chr1 10000 20000 ivl2
chr1 20000 30000 ivl3
chr1 30000 40000 ivl4
$ bedtools multicov -bams aln1.bam aln2.bam aln3.bam -bed ivls-of-interest.bed
chr1 0 10000 ivl1 100 2234 0
chr1 10000 20000 ivl2 123 3245 1000
chr1 20000 30000 ivl3 213 2332 2034
chr1 30000 40000 ivl4 335 7654 0
The output of multicov reflects a distinct report of the overlapping
alignments for each record in the -bed file. In the example above, each
line of the output reflects a) the original line from the -bed file
followed by b) the count of alignments that overlap the -bed interval
from each input -bam file. In the example above, the output consists of
7 columns: the first four of which are the columns from the -bed file and
the last 3 are the count of overlapping alignments from the 3 input -bam
files. The order of the counts reflects the order of the files given on the
command line.
Note
bedtools multicov will work with a single BAM as well.
$ bedtools multicov -bams aln1.bam -bed ivls-of-interest.bed
chr1 0 10000 ivl1 100
chr1 10000 20000 ivl2 123
chr1 20000 30000 ivl3 213
chr1 30000 40000 ivl4 335
multiinter¶

bedtools multiinteridentifies common intervals among multiple (and subsets thereof)- sorted BED/GFF/VCF files.
Note
- All files must be sorted in the same mmanner (e.g.,
sort -k 1,1 -k2,2n in.bed > in.sorted.bed)
Usage and option summary¶
Usage:
bedtools multiinter [OPTIONS] -i FILE1 FILE2 .. FILEn
(or):
multiIntersect [OPTIONS] -i FILE1 FILE2 .. FILEn
| Option | Description |
|---|---|
| -header | Print a header line (chrom/start/end + names of each file). |
| -names |
|
| -g | Use genome file to calculate empty regions. |
| -empty | Report empty regions (i.e., start/end intervals w/o
| values in all files). Requires the ‘-g FILE’ parameter.
|
| -filler TEXT | Use TEXT when representing intervals having no value.
| Default is ‘0’, but you can use ‘N/A’ or any text.
|
| -examples | Show usage examples on the command line. |
Default behavior¶
By default, bedtools multiinter will inspect all of the intervals in each input file and
report the sub-intervals that are overlapped by 0, 1, 2, … N files.
The default output format is as follows:
- chromosome (or entire genome)
- 0-based start coordinate of the sub-interval.
- 1-based end coordinate of the sub-interval.
- The number of files whose intervals overlap this sub interval at least once.
- The list of file numbers (by order on the command line) whose intervals overlap this sub interval at least once.
- Columns reflecting whether each file had (1) or did not have (0) 1 or more intervals overlapping this sub interval.
For example:
$ cat a.bed
chr1 6 12
chr1 10 20
chr1 22 27
chr1 24 30
cat b.bed
chr1 12 32
chr1 14 30
$ cat c.bed
chr1 8 15
chr1 10 14
chr1 32 34
$ cat sizes.txt
chr1 5000
$ bedtools multiinter -i a.bed b.bed c.bed
chr1 6 8 1 1 1 0 0
chr1 8 12 2 1,3 1 0 1
chr1 12 15 3 1,2,3 1 1 1
chr1 15 20 2 1,2 1 1 0
chr1 20 22 1 2 0 1 0
chr1 22 30 2 1,2 1 1 0
chr1 30 32 1 2 0 1 0
chr1 32 34 1 3 0 0 1
-header Add a header with columns names¶
For example:
$ bedtools multiinter -header -i a.bed b.bed c.bed
chrom start end num list a.bed b.bed c.bed
chr1 6 8 1 1 1 0 0
chr1 8 12 2 1,3 1 0 1
chr1 12 15 3 1,2,3 1 1 1
chr1 15 20 2 1,2 1 1 0
chr1 20 22 1 2 0 1 0
chr1 22 30 2 1,2 1 1 0
chr1 30 32 1 2 0 1 0
chr1 32 34 1 3 0 0 1
-names Add custom labels for each file in the header¶
For example:
$ bedtools multiinter -header -names A B C -i a.bed b.bed c.bed
chrom start end num list A B C
chr1 6 8 1 1 1 0 0
chr1 8 12 2 1,3 1 0 1
chr1 12 15 3 1,2,3 1 1 1
chr1 15 20 2 1,2 1 1 0
chr1 20 22 1 2 0 1 0
chr1 22 30 2 1,2 1 1 0
chr1 30 32 1 2 0 1 0
chr1 32 34 1 3 0 0 1
-empty Report the sub intervals not covered by any file¶
Note that this option requires a -g file so that it knows the full
range of each chromosome or contig.
For example:
$ bedtools multiinter -header -names A B C -i a.bed b.bed c.bed -empty -g sizes.txt
chrom start end num list A B C
chr1 0 6 0 none 0 0 0
chr1 6 8 1 A 1 0 0
chr1 8 12 2 A,C 1 0 1
chr1 12 15 3 A,B,C 1 1 1
chr1 15 20 2 A,B 1 1 0
chr1 20 22 1 B 0 1 0
chr1 22 30 2 A,B 1 1 0
chr1 30 32 1 B 0 1 0
chr1 32 34 1 C 0 0 1
chr1 34 5000 0 none 0 0 0
nuc¶
overlap¶
overlap computes the amount of overlap (in the case of positive values) or distance (in the case of negative values) between feature coordinates occurring on the same input line and reports the result at the end of the same line. In this way, it is a useful method for computing custom overlap scores from the output of other BEDTools.
Usage and option summary¶
Usage:
overlap [OPTIONS] -i <input> -cols s1,e1,s2,e2
| Option | Description |
|---|---|
| -i | Input file. Use “stdin” for pipes. |
| -cols | Specify the columns (1-based) for the starts and ends of the features for which you’d like to compute the overlap/distance. The columns must be listed in the following order: start1,end1,start2,end2 |
Default behavior¶
The default behavior is to compute the amount of overlap between the features you specify based on the start and end coordinates. For example:
bedtools window -a A.bed -b B.bed -w 10
chr1 10 20 A chr1 15 25 B
chr1 10 20 C chr1 25 35 D
# Now let’s say we want to compute the number of base pairs of overlap # between the overlapping features from the output of windowBed.
bedtools windo -a A.bed -b B.bed -w 10 | overlap -i stdin -cols 2,3,6,7
chr1 10 20 A chr1 15 25 B 5
chr1 10 20 C chr1 25 35 D -5
pairtobed¶
pairtopair¶
pairToPair compares two BEDPE files in search of overlaps where each end of a BEDPE feature in A overlaps with the ends of a feature in B. For example, using pairToPair, one could screen for the exact same discordant paired-end alignment in two files. This could suggest (among other things) that the discordant pair suggests the same structural variation in each file/sample.
Usage and option summary¶
Usage:
pairToPair [OPTIONS] -a <BEDPE> -b <BEDPE>
| Option | Description |
|---|---|
| -a | BEDPE file A. Each feature in A is compared to B in search of overlaps. Use “stdin” if passing A with a UNIX pipe. |
| -b | BEDPE file B. Use “stdin” if passing B with a UNIX pipe. |
| -f | Minimum overlap required as a fraction of A. Default is 1E-9 (i.e. 1bp). |
| -is | Force “strandedness”. That is, only report hits in B that overlap A on the same strand. By default, overlaps are reported without respect to strand. |
| -type |
either Report overlaps if either ends of A overlap B.
|
Default behavior¶
By default, a BEDPE feature from A will be reported if both ends overlap a feature in the BEDPE B file. If strand information is present for the two BEDPE files, it will be further required that the overlaps on each end be on the same strand. This way, an otherwise overlapping (in terms of genomic locations) F/R alignment will not be matched with a R/R alignment.
Default: Report A if both ends overlaps B.
Chromosome ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
BEDPE/BAM A *****.................................*****
BED File B ^^^^^^^^ ^^^^^^
Result =====.................................=====
Default when strand information is present in both BEDPE files: Report A if both ends overlaps B on the same strands.
Chromosome ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
BEDPE A >>>>>.................................>>>>>
BEDPE B <<<<<.............................>>>>>
Result
BEDPE A >>>>>.................................>>>>>
BEDPE B >>>>>.............................>>>>>
Result >>>>>.................................>>>>>
-type neither Optional overlap requirements¶
Using then -type neither, pairToPair will only report A if neither end overlaps with a BEDPE feature in B.
-type neither: Report A only if neither end overlaps B.
Chromosome ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
BEDPE/BAM A *****.................................*****
BED File B ^^^^^^^^......................................^^^^^^
Result
BEDPE/BAM A *****.................................*****
BED File B ^^^^................................................^^^^^^
Result =====.................................=====
random¶

bedtools random will generate a random set of intervals in BED6 format. One
can specify both the number (-n) and the size (-l) of the intervals
that should be generated.
Usage and option summary¶
Usage:
bedtools random [OPTIONS] -g <GENOME>
(or): : :
randomBed [OPTIONS] -g <GENOME>
| Option | Description |
|---|---|
| -l | The length of the intervals to generate.
Default = 100 |
| -n | The number of intervals to generate.
Default = 1,000,000 |
| -seed | Supply an integer seed for the shuffling. This will allow feature shuffling experiments to be recreated exactly as the seed for the pseudo-random number generation will be constant. By default, the seed is chosen automatically. |
Default behavior¶
By default, bedtools random generate 1 million intervals of length 100
placed randomly in the genome specificed with -g.
$ bedtools random -g hg19.genome
chr2 87536758 87536858 1 100 -
chrX 46051735 46051835 2 100 +
chr18 5237041 5237141 3 100 -
chr12 45809998 45810098 4 100 +
chrX 42034890 42034990 5 100 -
chr10 77510935 77511035 6 100 -
chr3 39844278 39844378 7 100 -
chr6 101012700 101012800 8 100 +
chr12 38123482 38123582 9 100 +
chr7 88508598 88508698 10 100 -
$ bedtools random -g hg19.genome
chr3 141987850 141987950 1 100 +
chr5 137643331 137643431 2 100 +
chr2 155523858 155523958 3 100 -
chr5 147874094 147874194 4 100 +
chr1 71838335 71838435 5 100 -
chr8 71154323 71154423 6 100 -
chr2 133240474 133240574 7 100 +
chr9 131495427 131495527 8 100 +
chrX 125952943 125953043 9 100 +
chr3 59685545 59685645 10 100 +
-n Specify the number of intervals to generate.¶
The -n option allows one to override the default of generating 1 million intervals.
$ bedtools random -g hg19.genome -n 3
chr20 47975280 47975380 1 100 -
chr16 23381222 23381322 2 100 +
chr3 104913816 104913916 3 100 -
-l Specify the length of intervals to generate.¶
The -l option allows one to override the default interval length of 100bp.
$ bedtools random -g hg19.genome -l 5
chr9 54133731 54133736 1 5 +
chr1 235288830 235288835 2 5 -
chr8 26744718 26744723 3 5 +
chr3 187313616 187313621 4 5 -
chr11 88996846 88996851 5 5 -
chr13 84714855 84714860 6 5 -
chr13 10759738 10759743 7 5 -
chr6 122569739 122569744 8 5 +
chr17 50884025 50884030 9 5 -
chr11 38576901 38576906 10 5 +
-seed Defining a “seed” for the random interval creation.¶
bedtools random uses a pseudo-random number generator to permute the locations of BED features. Therefore, each run should produce a different result. This can be problematic if one wants to exactly recreate an experiment. By using the seed option, one can supply a custom integer seed for bedtools random. In turn, each execution of bedtools random with the same seed and input files should produce identical results.
$ bedtools random -g hg19.genome -seed 71346
chrY 23380696 23380796 1 100 -
chr14 94368315 94368415 2 100 +
chr14 45353323 45353423 3 100 -
chr14 100546766 100546866 4 100 -
chr12 43294368 43294468 5 100 -
chr1 141470585 141470685 6 100 -
chr10 31273665 31273765 7 100 +
chr5 19102979 19103079 8 100 +
chr3 116730634 116730734 9 100 -
chr3 101222965 101223065 10 100 -
# (same seed, thus same as above)
$ bedtools random -g hg19.genome -seed 71346
chrY 23380696 23380796 1 100 -
chr14 94368315 94368415 2 100 +
chr14 45353323 45353423 3 100 -
chr14 100546766 100546866 4 100 -
chr12 43294368 43294468 5 100 -
chr1 141470585 141470685 6 100 -
chr10 31273665 31273765 7 100 +
chr5 19102979 19103079 8 100 +
chr3 116730634 116730734 9 100 -
chr3 101222965 101223065 10 100 -
reldist¶
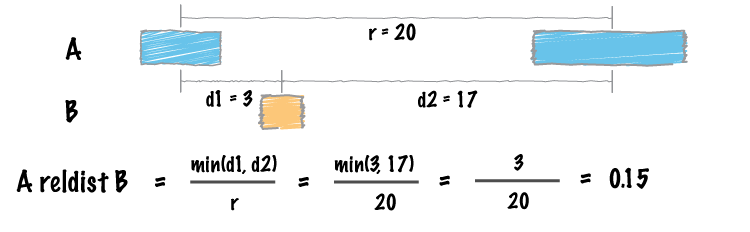
Traditional approaches to summarizing the similarity between two sets of genomic intervals are based upon the number or proportion of intersecting intervals. However, such measures are largely blind to spatial correlations between the two sets where, despite consistent spacing or proximity, intersections are rare (for example, enhancers and transcription start sites rarely overlap, yet they are much closer to one another than two sets of random intervals). Favorov et al [1] proposed a relative distance metric that describes distribution of relative distances between each interval in one set and the two closest intervals in another set (see figure above). If there is no spatial correlation between the two sets, one would expect the relative distances to be uniformly distributed among the relative distances ranging from 0 to 0.5. If, however, the intervals tend to be much closer than expected by chance, the distribution of observed relative distances would be shifted towards low relative distance values (e.g., the figure below).

[1] Exploring Massive, Genome Scale Datasets with the GenometriCorr Package.
Favorov A, Mularoni L, Cope LM, Medvedeva Y, Mironov AA, et al. (2012)
PLoS Comput Biol 8(5): e1002529. doi:10.1371/journal.pcbi.1002529
Usage and option summary¶
Usage:
bedtools reldist [OPTIONS] -a <BED/GFF/VCF> -b <BED/GFF/VCF>
| Option | Description |
|---|---|
| -a | BED/GFF/VCF file A. Each feature in A is compared to B in search of overlaps. Use “stdin” if passing A with a UNIX pipe. |
| -b | BED/GFF/VCF file B. Use “stdin” if passing B with a UNIX pipe. |
| -detail | Instead of a summary, report the relative distance for each interval in A |
Default behavior¶
By default, bedtools reldist reports the distribution of relative distances between two sets of intervals.
The output reports the frequency of each relative distance (ranging from 0.0 to 0.5). If the two sets of intervals
are randomly distributed with respect to one another, each relative distance “bin” with be roughly equally represented
(i.e., a uniform distribution). For example, consider the relative distance distance distribution for exons and AluY
elements:
$ bedtools reldist \
-a data/refseq.chr1.exons.bed.gz \
-b data/
aluY.chr1.bed.gz
0.00 164 43408 0.004
0.01 551 43408 0.013
0.02 598 43408 0.014
0.03 637 43408 0.015
0.04 793 43408 0.018
0.05 688 43408 0.016
0.06 874 43408 0.020
0.07 765 43408 0.018
0.08 685 43408 0.016
0.09 929 43408 0.021
0.10 876 43408 0.020
0.11 959 43408 0.022
0.12 860 43408 0.020
0.13 851 43408 0.020
0.14 903 43408 0.021
0.15 893 43408 0.021
0.16 883 43408 0.020
0.17 828 43408 0.019
0.18 917 43408 0.021
0.19 875 43408 0.020
0.20 897 43408 0.021
0.21 986 43408 0.023
0.22 903 43408 0.021
0.23 944 43408 0.022
0.24 904 43408 0.021
0.25 867 43408 0.020
0.26 943 43408 0.022
0.27 933 43408 0.021
0.28 1132 43408 0.026
0.29 881 43408 0.020
0.30 851 43408 0.020
0.31 963 43408 0.022
0.32 950 43408 0.022
0.33 965 43408 0.022
0.34 907 43408 0.021
0.35 884 43408 0.020
0.36 965 43408 0.022
0.37 944 43408 0.022
0.38 911 43408 0.021
0.39 939 43408 0.022
0.40 921 43408 0.021
0.41 950 43408 0.022
0.42 935 43408 0.022
0.43 919 43408 0.021
0.44 915 43408 0.021
0.45 934 43408 0.022
0.46 843 43408 0.019
0.47 850 43408 0.020
0.48 1006 43408 0.023
0.49 937 43408 0.022
In contrast, consider the relative distance distribution observed between exons and conserved elements:
$ bedtools reldist \
-a data/refseq.chr1.exons.bed.gz \
-b data/gerp.chr1.bed.gz
reldist count total fraction
0.00 20629 43422 0.475
0.01 2629 43422 0.061
0.02 1427 43422 0.033
0.03 985 43422 0.023
0.04 897 43422 0.021
0.05 756 43422 0.017
0.06 667 43422 0.015
0.07 557 43422 0.013
0.08 603 43422 0.014
0.09 487 43422 0.011
0.10 461 43422 0.011
0.11 423 43422 0.010
0.12 427 43422 0.010
0.13 435 43422 0.010
0.14 375 43422 0.009
0.15 367 43422 0.008
0.16 379 43422 0.009
0.17 371 43422 0.009
0.18 346 43422 0.008
0.19 389 43422 0.009
0.20 377 43422 0.009
0.21 411 43422 0.009
0.22 377 43422 0.009
0.23 352 43422 0.008
0.24 334 43422 0.008
0.25 315 43422 0.007
0.26 370 43422 0.009
0.27 330 43422 0.008
0.28 330 43422 0.008
0.29 280 43422 0.006
0.30 309 43422 0.007
0.31 326 43422 0.008
0.32 287 43422 0.007
0.33 294 43422 0.007
0.34 306 43422 0.007
0.35 307 43422 0.007
0.36 309 43422 0.007
0.37 271 43422 0.006
0.38 293 43422 0.007
0.39 311 43422 0.007
0.40 331 43422 0.008
0.41 320 43422 0.007
0.42 299 43422 0.007
0.43 327 43422 0.008
0.44 321 43422 0.007
0.45 326 43422 0.008
0.46 306 43422 0.007
0.47 354 43422 0.008
0.48 365 43422 0.008
0.49 336 43422 0.008
0.50 38 43422 0.001
Moreover, if one compares the relative distances for one set against itself, every interval should be expected to overlap an interval in the other set (itself). As such, the relative distances will all be 0.0:
$ bedtools reldist \
-a data/refseq.chr1.exons.bed.gz \
-b data/refseq.chr1.exons.bed.gz
reldist count total fraction
0.00 43424 43424 1.000
shift¶

bedtools shift will move each feature in a feature file by a
user-defined number of bases. While something like this could be done with an
awk '{OFS="\t" print $1,$2+<shift>,$3+<shift>}',
bedtools shift will restrict the resizing to the size of the chromosome
(i.e. no features before 0 or past the chromosome end).
Note
In order to prevent the extension of intervals beyond chromosome boundaries,
bedtools shift requires a genome file defining the length of each
chromosome or contig.
See also
Usage and option summary¶
Usage:
bedtools shift [OPTIONS] -i <BED/GFF/VCF> -g <GENOME> [-s or (-m and -p)]
(or):
shiftBed [OPTIONS] -i <BED/GFF/VCF> -g <GENOME> [-s or (-m and -p)]
| Option | Description |
|---|---|
| -s | Shift the BED/GFF/VCF entry -s base pairs. Integer or Float (e.g. 0.1) if used with -pct |
| -m | Shift entries on the - strand -m base pairs. Integer or Float (e.g. 0.1) if used with -pct |
| -p | Shift entries on the + strand -p base pairs. Integer or Float (e.g. 0.1) if used with -pct |
| -pct | Define -s, -m and -p as a fraction of the feature’s length. E.g. if used on a 1000bp feature, -s 0.50, will shift the feature 500 bp “upstream”. Default = false. |
| -header | Print the header from the input file prior to results. |
Default behavior¶
By default, bedtools shift will shift features a fixed number of bases. This shift can either be applied to all features (-s), or seperately to features on the plus (-p) and minus (-m) strands. Shifts can either be positive or negative.
$ cat A.bed
chr1 5 100 +
chr1 800 980 -
$ cat my.genome
chr1 1000
$ bedtools shift -i A.bed -g my.genome -s 5
chr1 10 105 +
chr1 805 985 -
$ bedtools shift -i A.bed -g my.genome -p 2 -m -3
chr1 7 102 +
chr1 797 977 -
However, if the requested number of bases exceeds the boundaries of the
chromosome, bedtools shift will “clip” the feature accordingly.
$ cat A.bed
chr1 5 100 +
chr1 800 980 +
$ cat my.genome
chr1 1000
$ bedtools shift -i A.bed -g my.genome -s 5000
chr1 999 1000 +
chr1 999 1000 +
-pct Shifting features by a given fraction¶
bedtools shift will shift the feature by a user-specific fraction of the
feature length. Hence, 0.5 will add half the length of the feature to the start and end coordinates.
For example:
$ cat A.bed
chr1 100 200 a1 1 +
$ bedtools shift -i A.bed -g my.genome -s 0.5 -pct
chr1 150 250 a1 1 +
-header Print the header for the A file before reporting results.¶
By default, if your A file has a header, it is ignored when reporting results. This option will instead tell bedtools to first print the header for the A file prior to reporting results.
shuffle¶
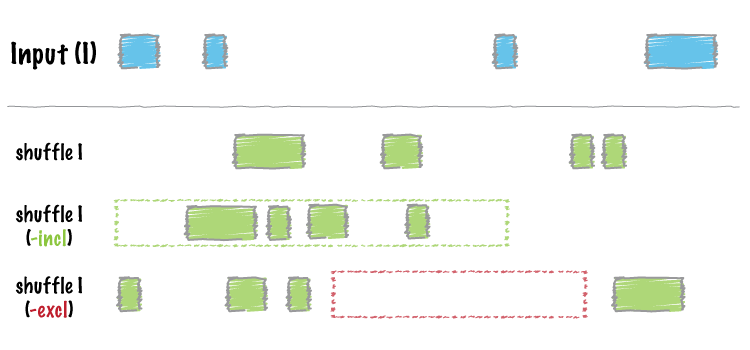
bedtools shuffle will randomly permute the genomic locations of a feature file among a genome defined in a genome file. One can also provide an “exclusions” BED/GFF/VCF file that lists regions where you do not want the permuted features to be placed. For example, one might want to prevent features from being placed in known genome gaps. shuffle is useful as a null basis against which to test the significance of associations of one feature with another.
Usage and option summary¶
Usage:
bedtools shuffle [OPTIONS] -i <BED/GFF/VCF> -g <GENOME>
(or):
shuffleBed [OPTIONS] -i <BED/GFF/VCF> -g <GENOME>
| Option | Description |
|---|---|
| -excl | A BED file of coordinates in which features from -i should not be placed (e.g., genome gaps). |
| -incl | A BED file of coordinates in which features from -i should be placed. |
| -chrom | Keep features in -i on the same chromosome. Solely permute their location on the chromosome. By default, both the chromosome and position are randomly chosen. |
| -seed | Supply an integer seed for the shuffling. This will allow feature shuffling experiments to be recreated exactly as the seed for the pseudo-random number generation will be constant. By default, the seed is chosen automatically. |
| -f | Maximum overlap (as a fraction of the -i feature) with an -excl feature that is tolerated before searching for a new, randomized locus. |
| -chromFirst | Instead of choosing a position randomly among the entire genome (the default), first choose a chrom randomly, and then choose a random start coordinate on that chrom. This leads to features being ~uniformly distributed among the chroms, as opposed to features being distribute as a function of chrom size. |
| -bedpe | Indicate that the A file is in BEDPE format. |
| -maxTries | Max. number of attempts to find a home for a shuffled interval in the presence of -incl or -excl. Default = 1000. |
| -noOverlapping | Don’t allow shuffled intervals to overlap. |
| -allowBeyondChromEnd | Allow the original the length of the original records to extend beyond the length of the chromosome. |
Default behavior¶
By default, bedtools shuffle will reposition each feature in the input BED file on a random chromosome at a random position. The size and strand of each feature are preserved.
For example:
$ cat A.bed
chr1 0 100 a1 1 +
chr1 0 1000 a2 2 -
$ cat my.genome
chr1 10000
chr2 8000
chr3 5000
chr4 2000
$ bedtools shuffle -i A.bed -g my.genome
chr4 1498 1598 a1 1 +
chr3 2156 3156 a2 2 -
-chrom Requiring that features be shuffled on the same chromosome¶
The -chrom option behaves the same as the default behavior except that features are randomly placed on the same chromosome as defined in the BED file.
$ cat A.bed
chr1 0 100 a1 1 +
chr1 0 1000 a2 2 -
$ cat my.genome
chr1 10000
chr2 8000
chr3 5000
chr4 2000
$ bedtools shuffle -i A.bed -g my.genome -chrom
chr1 9560 9660 a1 1 +
chr1 7258 8258 a2 2 -
-excl Excluding certain genome regions from bedtools shuffle¶
One may want to prevent BED features from being placed in certain regions of
the genome. For example, one may want to exclude genome gaps from permutation
experiment. The excl option defines a BED file of regions that should be
excluded. bedtools shuffle will attempt to permute the locations of all features
while adhering to the exclusion rules. However it will stop looking for an
appropriate location if it cannot find a valid spot for a feature
after 1,000,000 tries.
For example (note that the exclude file excludes all but 100 base pairs of the chromosome):
$ cat A.bed
chr1 0 100 a1 1 +
chr1 0 1000 a2 2 -
$ cat my.genome
chr1 10000
$ cat exclude.bed
chr1 100 10000
$ bedtools shuffle -i A.bed -g my.genome -excl exclude.bed
chr1 0 100 a1 1 +
Error, line 2: tried 1000000 potential loci for entry, but could not avoid excluded
regions. Ignoring entry and moving on.
For example (now the exclusion file only excludes the first 100 bases of the chromosome):
$ cat A.bed
chr1 0 100 a1 1 +
chr1 0 1000 a2 2 -
$ cat my.genome
chr1 10000
$ cat exclude.bed
chr1 0 100
$ bedtools shuffle -i A.bed -g my.genome -excl exclude.bed
chr1 147 247 a1 1 +
chr1 2441 3441 a2 2 -
-seed Defining a “seed” for the random replacement.¶
bedtools shuffle uses a pseudo-random number generator to permute the locations of BED features. Therefore, each run should produce a different result. This can be problematic if one wants to exactly recreate an experiment. By using the seed option, one can supply a custom integer seed for bedtools shuffle. In turn, each execution of bedtools shuffle with the same seed and input files should produce identical results.
For example (note that the exclude file below excludes all but 100 base pairs of the chromosome):
$ cat A.bed
chr1 0 100 a1 1 +
chr1 0 1000 a2 2 -
$ cat my.genome
chr1 10000
$ bedtools shuffle -i A.bed -g my.genome -seed 927442958
chr1 6177 6277 a1 1 +
chr1 8119 9119 a2 2 -
$ bedtools shuffle -i A.bed -g my.genome -seed 927442958
chr1 6177 6277 a1 1 +
chr1 8119 9119 a2 2 -
. . .
$ bedtools shuffle -i A.bed -g my.genome -seed 927442958
chr1 6177 6277 a1 1 +
chr1 8119 9119 a2 2 -
-noOverlapping Prevent shuffled intervals from overlapping.¶
There often arise cases where one wants to shuffle intervals throughout
the genome, yet one wants to prevent the intervals from occupying a single
common base pair. The -noOverlapping option allows one to enforce
no such overlaps.
$ bedtools shuffle -i A.bed -g my.genome -noOverlapping
-allowBeyondChromEnd Allow records to extend beyond the chrom. length.¶
By default, shuffle requires that an interval’s original length must be
fully-contained within the chromosome. Yet there are cases where you might
want to allow shuffled intervals to be relocated to a position
in which the entire original interval cannot fit w/o exceeding
the end of the chromosome. By using the -noOverlapping option,
shuffle will allow intervals to be shuffled to locations that are so close
to the chromosome end that the full length of the original record cannot
be contained within the chromosome length. In such cases, the end coordinate
for the shuffled interval will be set (i.e., truncated)
to the chromosome’s length.
$ bedtools shuffle -i A.bed -g my.genome -allowBeyondChromEnd
slop¶
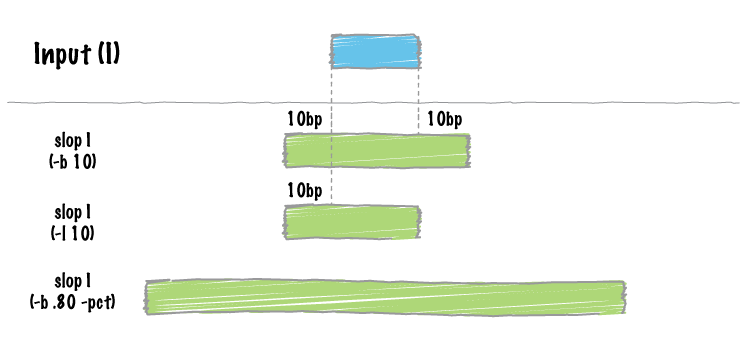
bedtools slop will increase the size of each feature in a feature file by a
user-defined number of bases. While something like this could be done with an
awk '{OFS="\t" print $1,$2-<slop>,$3+<slop>}',
bedtools slop will restrict the resizing to the size of the chromosome
(i.e. no start < 0 and no end > chromosome size).
Note
In order to prevent the extension of intervals beyond chromosome boundaries,
bedtools slop requires a genome file defining the length of each
chromosome or contig.
See also
Usage and option summary¶
Usage:
bedtools slop [OPTIONS] -i <BED/GFF/VCF> -g <GENOME> [-b or (-l and -r)]
(or):
slopBed [OPTIONS] -i <BED/GFF/VCF> -g <GENOME> [-b or (-l and -r)]
| Option | Description |
|---|---|
| -b | Increase the BED/GFF/VCF entry by the same number base pairs in each direction. Integer. |
| -l | The number of base pairs to subtract from the start coordinate. Integer. |
| -r | The number of base pairs to add to the end coordinate. Integer. |
| -s | Define -l and -r based on strand. For example. if used, -l 500 for a negative-stranded feature, it will add 500 bp to the end coordinate. |
| -pct | Define -l and -r as a fraction of the feature’s length. E.g. if used on a 1000bp feature, -l 0.50, will add 500 bp “upstream”. Default = false. |
| -header | Print the header from the input file prior to results. |
Default behavior¶
By default, bedtools slop will either add a fixed number of bases in each
direction (-b) or an asymmetric number of bases in each direction
with -l and -r.
$ cat A.bed
chr1 5 100
chr1 800 980
$ cat my.genome
chr1 1000
$ bedtools slop -i A.bed -g my.genome -b 5
chr1 0 105
chr1 795 985
$ bedtools slop -i A.bed -g my.genome -l 2 -r 3
chr1 3 103
chr1 798 983
However, if the requested number of bases exceeds the boundaries of the
chromosome, bedtools slop will “clip” the feature accordingly.
$ cat A.bed
chr1 5 100
chr1 800 980
$ cat my.genome
chr1 1000
$ bedtools slop -i A.bed -g my.genome -b 5000
chr1 0 1000
chr1 0 1000
-s Resizing features according to strand¶
bedtools slop will optionally increase the size of a feature based on strand.
For example:
$ cat A.bed
chr1 100 200 a1 1 +
chr1 100 200 a2 2 -
$ cat my.genome
chr1 1000
$ bedtools slop -i A.bed -g my.genome -l 50 -r 80 -s
chr1 50 280 a1 1 +
chr1 20 250 a2 2 -
-pct Resizing features by a given fraction¶
bedtools slop will optionally increase the size of a feature by a
user-specific fraction.
For example:
$ cat A.bed
chr1 100 200 a1 1 +
$ bedtools slop -i A.bed -g my.genome -b 0.5 -pct
chr1 50 250 a1 1 +
$ bedtools slop -i a.bed -l 0.5 -r 0.0 -pct -g my.genome
chr1 50 200 a1 1 +
-header Print the header for the A file before reporting results.¶
By default, if your A file has a header, it is ignored when reporting results. This option will instead tell bedtools to first print the header for the A file prior to reporting results.
sort¶
The bedtools sort tool sorts a feature file by chromosome and other criteria.
Usage and option summary¶
Usage:
bedtools sort [OPTIONS] -i <BED/GFF/VCF>
(or):
sortBed [OPTIONS] -i <BED/GFF/VCF>
| Option | Description |
|---|---|
| -sizeA | Sort by feature size in ascending order. |
| -sizeD | Sort by feature size in descending order. |
| -chrThenSizeA | Sort by chromosome (asc), then by feature size (asc). |
| -chrThenSizeD | Sort by chromosome (asc), then by feature size (desc). |
| -chrThenScoreA | Sort by chromosome (asc), then by score (asc). |
| -chrThenScoreD | Sort by chromosome (asc), then by score (desc). |
| -g | Define sort order by order of tab-delimited file with chromosome names in the first column. |
| -faidx | Define sort order by order of tab-delimited file with chromosome names in the first column. Sort by specified chromosome order. |
Default behavior¶
By default, bedtools sort sorts a BED file by chromosome and then by start position in ascending order.
For example:
cat A.bed
chr1 800 1000
chr1 80 180
chr1 1 10
chr1 750 10000
sortBed -i A.bed
chr1 1 10
chr1 80 180
chr1 750 10000
chr1 800 1000
Optional sorting behavior¶
bedtools sort will also sort a BED file by chromosome and then by other criteria.
For example, to sort by chromosome and then by feature size (in descending order):
cat A.bed
chr1 800 1000
chr1 80 180
chr1 1 10
chr1 750 10000
sortBed -i A.bed -sizeD
chr1 750 10000
chr1 800 1000
chr1 80 180
chr1 1 10
Disclaimer: it should be noted that bedtools sort is merely a convenience utility, as the UNIX sort utility
will sort BED files more quickly while using less memory. For example, UNIX sort will sort a BED file
by chromosome then by start position in the following manner:
sort -k 1,1 -k2,2n a.bed
chr1 1 10
chr1 80 180
chr1 750 10000
chr1 800 1000
subtract¶
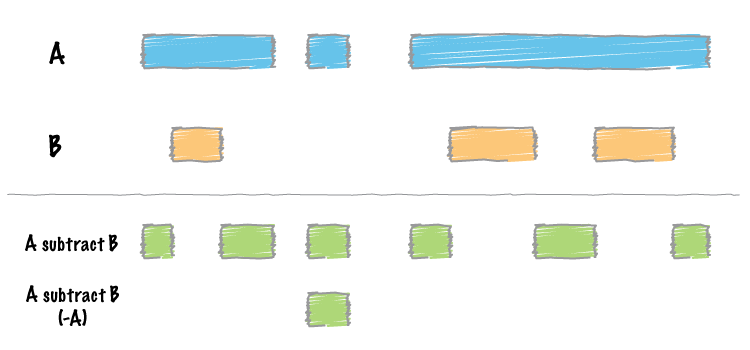
bedtools subtract bedtools` subtract searches for features in B that overlap A by at least the number of base pairs given by the -f option. If an overlapping feature is found in B, the overlapping portion is removed from A and the remaining portion of A is reported. If a feature in B overlaps all of a feature in A, the A feature will not be reported. If a feature in B does not overlap a feature in A by at least the -f amount, the A feature will be reported in its entirety.
Usage and option summary¶
Usage:
bedtools subtract [OPTIONS] -a <BED/GFF/VCF> -b <BED/GFF/VCF>
(or):
subtractBed [OPTIONS] -a <BED/GFF/VCF> -b <BED/GFF/VCF>
| Option | Description |
|---|---|
| -f | Minimum overlap required as a fraction of A. Default is 1E-9 (i.e. 1bp). |
| -F | Minimum overlap required as a fraction of B. Default is 1E-9 (i.e., 1bp). |
| -r | Require that the fraction of overlap be reciprocal for A and B. In other words, if -f is 0.90 and -r is used, this requires that B overlap at least 90% of A and that A also overlaps at least 90% of B. |
| -e | Require that the minimum fraction be satisfied for A _OR_ B. In other words, if -e is used with -f 0.90 and -F 0.10 this requires that either 90% of A is covered OR 10% of B is covered. Without -e, both fractions would have to be satisfied. |
| -s | Force “strandedness”. That is, only report hits in B that overlap A on the same strand. By default, overlaps are reported without respect to strand. |
| -S | Require different strandedness. That is, only report hits in B that overlap A on the _opposite_ strand. By default, overlaps are reported without respect to strand. |
| -A | Remove entire feature if any overlap. That is, by default, only subtract the portion of A that overlaps B. Here, if any overlap is found (or -f amount), the entire feature is removed. |
| -N | Same as -A except when used with -f, the amount is the sum of all features (not any single feature). |
Default behavior¶
By default, bedtools subtracts removes each overlapping interval in B
from A. If a feature in B completely overlaps a feature in A, the A feature
is removed.
$ cat A.bed
chr1 10 20
chr1 100 200
$ cat B.bed
chr1 0 30
chr1 180 300
$ bedtools subtract -a A.bed -b B.bed
chr1 100 180
-f Requiring a minimal overlap fraction before subtracting¶
This option behaves the same as the -f option for bedtools intersect.
In this case, subtract will only subtract an overlap with B if it covers at
least the fraction of A defined by -f. If an overlap is found,
but it does not meet the overlap fraction, the original A feature is
reported without subtraction.
$ cat A.bed
chr1 100 200
$ cat B.bed
chr1 180 300
$ bedtools subtract -a A.bed -b B.bed -f 0.10
chr1 100 180
$ bedtools subtract -a A.bed -b B.bed -f 0.80
chr1 100 200
-s Enforcing same “strandedness”¶
This option behaves the same as the -s option for bedtools intersect
while scanning for features in B that should be subtracted from A.
$ cat A.bed
chr1 100 200 a1 1 +
$ cat B.bed
chr1 80 120 b1 1 +
chr1 180 300 b2 1 -
$ bedtools subtract -a A.bed -b B.bed -s
chr1 120 200 a1 1 +
-S Enforcing opposite “strandedness”¶
This option behaves the same as the -s option for bedtools intersect
while scanning for features in B that should be subtracted from A.
$ cat A.bed
chr1 100 200 a1 1 +
$ cat B.bed
chr1 80 120 b1 1 +
chr1 180 300 b2 1 -
$ bedtools subtract -a A.bed -b B.bed -S
chr1 100 180 a1 1 +
-A Remove features with any overlap¶
Unlike the default behavior, the -A option will completely remove
a feature from A if it has even 1bp of overlap with a feature in B.
$ cat A.bed
chr1 100 200
$ cat B.bed
chr1 180 300
$ bedtools subtract -a A.bed -b B.bed
chr1 100 180
$ bedtools subtract -a A.bed -b B.bed -A
summary¶
Genomics experiments have numerous sources of both technical and biological variation
that can confound analysis and interpretation. Therefore, one of the most important steps
in genomics data analysis is generating high-level summary stats of one’s data to ask if the
basic observations align with the expectation. Doing such quality control as early
as possible in the analysis workflow helps to head off unnecessary time spent chasing
technical artifacts that masquerade as biological signals. This quality control
is the motivation behind the bedtools summary command.
Given an input interval file in standard formats, as well as a genome file defining
the chromosome names and lengths relevant to your data, bedtools summary will compute
several summary statistics detailing, for each chromosome, the number of intervals,
the total number of base pairs, and the fraction of intervals and base pairs observed
in your input file. From these summary measures, one can get a quick sense of questions like:
- Are all chromosomes represented in my data or annotation file?
- Do all chromosomes have the expected number of intervals or fraction of base pairs represented?
- Which chromosomes are outliers?
For example, the following plot was generated directly from the output of bedtools summary.
It depicts, for each chromosome, the fraction of all intervals in the RepeatMasker track from UCSC
observed on each chromosome versus the fraction of RepeatMasker intervals that are expected
for each chromosome, based on the fraction of the genome that each chromosome represents.
This plot highlights that chr19, chrM, and chrMT (the different mitochondrial reference genomes) are outliers.
Chromosome 19 has more than 1.5 times the intervals that are expected based upon the length of
the chromosome. ChrM has no intervals, making the observed-to-expected ratio be 0.
This former is because the repeat content of chromosome 19 “is approximately 55%, more than 10% higher
than the genome-wide average” (Grimwood J, et al. Nature. 2004;428:529–35). The latter is because
are indeed no repeat annotations provided by UCSC for the mitochondrial genome.
In this case, the extremes in observed versus expected ratios make sense. However, this tool allows one to detect cases that do not and are either artifacts or biological signals.
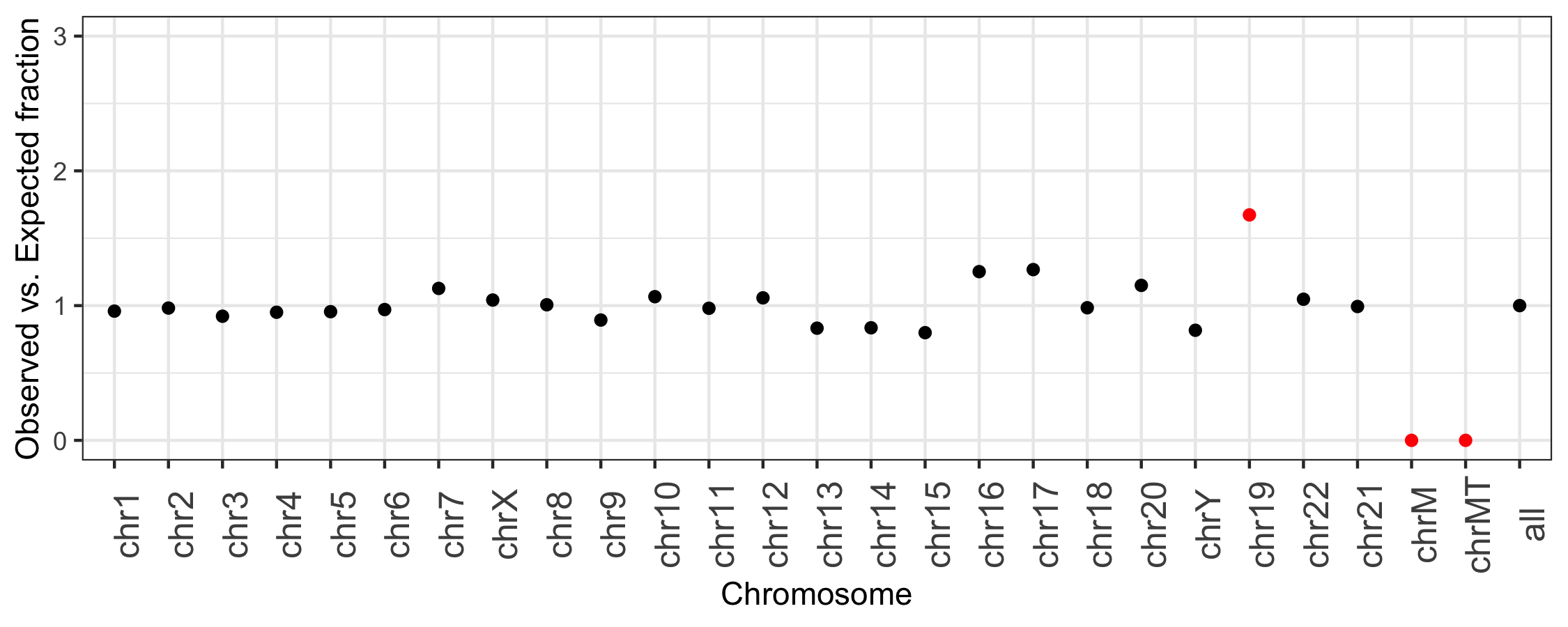
Default behavior¶
bedtools summary scans the input interval file and the chromosome lengths provided in
the “genome” file and reports the following columns for each chromosome.
- chrom (chromosome name)
- chrom_length (the length of the chromosome in bp)
- num_ivls (the total number of intervals observed for the chromosome)
- total_ivl_bp (the total number of bp observed in the intervals for the chromosome)
- chrom_frac_genome (the fraction of the genome represented by the chromosome)
- frac_all_ivls (the fraction of all intervals observed the chromosome)
- frac_all_bp (the fraction of all bp observed the chromosome)
- min (the smallest interval observed on that chromosome)
- max (the largest interval observed on that chromosome)
- mean (the mean interval length observed on that chromosome)
In addition, bedtools summary reports a final line with the chromosome name “all”, which
depicts the metrics above but tabulated across all chromosomes in the input file.
As an example, let’s recreate the figure above by running bedtools summary on the
simple repeats track from UCSC. We’ll begin by downloading the data in track the
track table and converting it to BED format.
curl -s http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/simpleRepeat.txt.gz \
| gzcat \
| cut -f 2-5 \
| grep -v -E 'Un|fix|random|alt|hap' \
> simrep.grch38.bed
head simrep.grch38.bed
chr1 10000 10468 trf
chr1 10627 10800 trf
chr1 10757 10997 trf
chr1 11225 11447 trf
chr1 11271 11448 trf
chr1 11283 11448 trf
chr1 19305 19443 trf
chr1 20828 20863 trf
chr1 30862 30959 trf
chr1 44835 44876 trf
Now, let’s make a “genome” file for GRCh38 from the chromInfo table at UCSC
curl -s http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/chromInfo.txt.gz \
| gzcat \
| cut -f 1-2 \
| grep -v -E 'Un|fix|random|alt|hap' \
> grch38.genome.txt
head grch38.genome.txt
chr1 248956422
chr2 242193529
chr3 198295559
chr4 190214555
chr5 181538259
chr6 170805979
chr7 159345973
chrX 156040895
chr8 145138636
chr9 138394717
Now, let’s run bedtools summary.
bedtools summary -i simrep.grch38.bed -g grch38.genome.txt | column -t
chrom chrom_length num_ivls total_ivl_bp chrom_frac_genome frac_all_ivls frac_all_bp min max mean
chr1 248956422 74548 15557884 0.080613126 0.077210928 0.048725518 25 124438 208.696195740
chr2 242193529 74474 14493548 0.078423273 0.077134284 0.045392139 25 336509 194.612186803
chr3 198295559 56894 13946854 0.064208928 0.058926309 0.043679955 25 500000 245.137518895
chr4 190214555 56685 10160257 0.061592265 0.058709844 0.031820766 25 136950 179.240663315
chr5 181538259 53887 16801740 0.058782844 0.055811896 0.052621133 25 500000 311.795794904
chr6 170805979 51802 11222841 0.055307687 0.053652418 0.035148658 25 500000 216.648797344
chr7 159345973 55972 20054618 0.051596890 0.057971375 0.062808775 25 150228 358.297327235
chrX 156040895 50432 27398336 0.050526692 0.052233481 0.085808462 25 500000 543.272842640
chr8 145138636 45937 15650021 0.046996495 0.047577915 0.049014080 25 500000 340.684437382
chr9 138394717 39329 10932158 0.044812786 0.040733870 0.034238272 25 159861 277.966843805
chr11 135086622 41279 14127024 0.043741611 0.042753526 0.044244227 25 500000 342.232709126
chr10 133797422 45074 11407694 0.043324163 0.046684087 0.035727596 25 110000 253.088121755
chr12 133275309 44151 13878240 0.043155100 0.045728117 0.043465064 25 356015 314.335802134
chr13 114364328 29907 9423815 0.037031646 0.030975307 0.029514313 25 110000 315.103989033
chr14 107043718 27973 9245970 0.034661202 0.028972223 0.028957323 25 173523 330.531941515
chr15 101991189 25557 9565023 0.033025172 0.026469921 0.029956561 25 110000 374.262354736
chr16 90338345 35288 11959674 0.029251932 0.036548522 0.037456334 25 138208 338.916175470
chr17 83257441 32093 16264416 0.026959106 0.033239393 0.050938295 25 132210 506.790141152
chr18 80373285 23966 18684937 0.026025204 0.024822089 0.058519091 25 500000 779.643536677
chr20 64444167 22608 12620160 0.020867290 0.023415580 0.039524901 25 500000 558.216560510
chr19 58617616 30854 11391752 0.018980628 0.031956135 0.035677667 25 396802 369.214753355
chrY 57227415 15130 4564760 0.018530475 0.015670458 0.014296307 25 227093 301.702577660
chr22 50818468 16760 10691540 0.016455232 0.017358684 0.033484683 25 498537 637.920047733
chr21 46709983 14911 9253172 0.015124887 0.015443636 0.028979879 25 499939 620.560123399
chrM 16569 0 0 0.000005365 0.000000000 0.000000000 -1 -1 -1
all 3088286401 965511 319296434 1.0 1.0 1.0 25 500000 330.702015824
Notice the following:
- There are 0 intervals reported for chrM or chrMT; therefore, the min, max, and mean are all “-1”.
- The last line in the output is has an “genome” chromosome, meaning it is a summary of all of the chromosomes.
Using this report, there are many high-level sanity checks one can explore. For example, we can create the plot described above by saving the output to a file.
bedtools summary -i simrep.grch38.bed -g grch38.genome.txt > ~/simrep.summary.tsv
Now run following R code. (Sorry, I am not an R expert)
if (!require("dplyr")) install.packages("dplyr")
if (!require("ggplot2")) install.packages("ggplot2")
library(dplyr)
library(ggplot2)
x = read_tsv('~/simrep.summary.tsv')
x = x %>% mutate(obs_v_exp = frac_all_ivls/chrom_frac_genome)
p = ggplot(x) +
ylim(0,3) +
ylab("Observed vs. Expected fraction") +
xlab("Chromosome") +
geom_point(aes(x=factor(chrom, level=chrom),
y=obs_v_exp,
color=ifelse(obs_v_exp>1.5 | obs_v_exp<0.5, 'red', 'black'))) +
scale_color_identity() +
theme_bw()
p + theme(axis.text.x = element_text(size = 12, angle = 90))
tag¶
unionbedg¶
bedtools unionbedg combines multiple BEDGRAPH files into a single file such that one can directly compare coverage (and other text-values such as genotypes) across multiple sample
Usage and option summary¶
Usage:
bedtools unionbedg [OPTIONS] -i FILE1 FILE2 FILE3 ... FILEn
| Option | Description |
|---|---|
| -header | Print a header line, consisting of chrom, start, end followed by the names of each input BEDGRAPH file. |
| -names | A list of names (one per file) to describe each file in -i. These names will be printed in the header line. |
| -empty | Report empty regions (i.e., start/end intervals w/o values in all files). Requires the ‘-g FILE’ parameter (see below). |
| -g | The genome file to be used to calculate empty regions. |
| -filler TEXT | Use TEXT when representing intervals having no value. Default is ‘0’, but you can use ‘N/A’ or any other text. |
| -examples | Show detailed usage examples. |
Default behavior¶
cat 1.bg
chr1 1000 1500 10
chr1 2000 2100 20
cat 2.bg
chr1 900 1600 60
chr1 1700 2050 50
cat 3.bg
chr1 1980 2070 80
chr1 2090 2100 20
cat sizes.txt
chr1 5000
bedtools unionbedg -i 1.bg 2.bg 3.bg
chr1 900 1000 0 60 0
chr1 1000 1500 10 60 0
chr1 1500 1600 0 60 0
chr1 1700 1980 0 50 0
chr1 1980 2000 0 50 80
chr1 2000 2050 20 50 80
chr1 2050 2070 20 0 80
chr1 2070 2090 20 0 0
chr1 2090 2100 20 0 20
-header Add a header line to the output¶
bedtools unionbedg -i 1.bg 2.bg 3.bg -header
chrom start end 1 2 3
chr1 900 1000 0 60 0
chr1 1000 1500 10 60 0
chr1 1500 1600 0 60 0
chr1 1700 1980 0 50 0
chr1 1980 2000 0 50 80
chr1 2000 2050 20 50 80
chr1 2050 2070 20 0 80
chr1 2070 2090 20 0 0
chr1 2090 2100 20 0 20
-names Add a header line with custom file names to the output¶
bedtools unionbedg -i 1.bg 2.bg 3.bg -header -names WT-1 WT-2 KO-1
chrom start end WT-1 WT-2 KO-1
chr1 900 1000 0 60 0
chr1 1000 1500 10 60 0
chr1 1500 1600 0 60 0
chr1 1700 1980 0 50 0
chr1 1980 2000 0 50 80
chr1 2000 2050 20 50 80
chr1 2050 2070 20 0 80
chr1 2070 2090 20 0 0
chr1 2090 2100 20 0 20
-empty Include regions that have zero coverage in all BEDGRAPH files.¶
bedtools unionbedg -i 1.bg 2.bg 3.bg -empty -g sizes.txt -header
chrom start end WT-1 WT-2 KO-1
chrom start end 1 2 3
chr1 0 900 0 0 0
chr1 900 1000 0 60 0
chr1 1000 1500 10 60 0
chr1 1500 1600 0 60 0
chr1 1600 1700 0 0 0
chr1 1700 1980 0 50 0
chr1 1980 2000 0 50 80
chr1 2000 2050 20 50 80
chr1 2050 2070 20 0 80
chr1 2070 2090 20 0 0
chr1 2090 2100 20 0 20
chr1 2100 5000 0 0 0
-filler Use a custom value for missing values.¶
bedtools unionbedg -i 1.bg 2.bg 3.bg -empty -g sizes.txt -header -filler N/A
chrom start end WT-1 WT-2 KO-1
chrom start end 1 2 3
chr1 0 900 N/A N/A N/A
chr1 900 1000 N/A 60 N/A
chr1 1000 1500 10 60 N/A
chr1 1500 1600 N/A 60 N/A
chr1 1600 1700 N/A N/A N/A
chr1 1700 1980 N/A 50 N/A
chr1 1980 2000 N/A 50 80
chr1 2000 2050 20 50 80
chr1 2050 2070 20 N/A 80
chr1 2070 2090 20 N/A N/A
chr1 2090 2100 20 N/A 20
chr1 2100 5000 N/A N/A N/A
Use BEDGRAPH files with non-numeric values.¶
cat 1.snp.bg
chr1 0 1 A/G
chr1 5 6 C/T
cat 2.snp.bg
chr1 0 1 C/C
chr1 7 8 T/T
cat 3.snp.bg
chr1 0 1 A/G
chr1 5 6 C/T
bedtools unionbedg -i 1.snp.bg 2.snp.bg 3.snp.bg -filler -/-
chr1 0 1 A/G C/C A/G
chr1 5 6 C/T -/- C/T
chr1 7 8 -/- T/T -/-
window¶
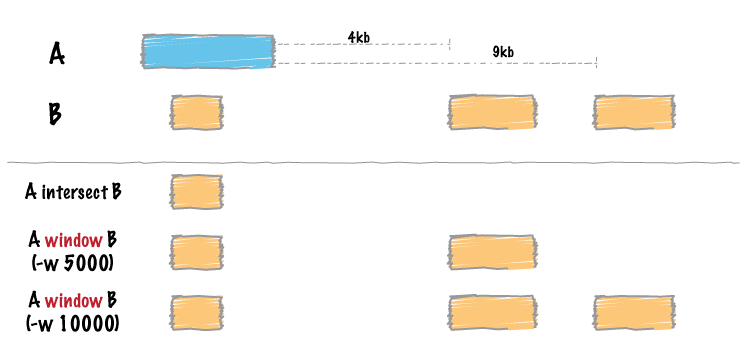
Similar to bedtools intersect, window searches for overlapping features
in A and B. However, window adds a specified number (1000, by default) of
base pairs upstream and downstream of each feature in A. In effect, this allows
features in B that are “near” features in A to be detected.
Usage and option summary¶
Usage:
bedtools window [OPTIONS] [-a|-abam] -b <BED/GFF/VCF>
(or):
bedtools window [OPTIONS] [-a|-abam] -b <BED/GFF/VCF>
| Option | Description |
|---|---|
| -abam | BAM file A. Each BAM alignment in A is compared to B in search of overlaps. Use “stdin” if passing A with a UNIX pipe: For example: samtools view -b <BAM> | bedtools window -abam stdin -b genes.bed |
| -ubam | Write uncompressed BAM output. The default is write compressed BAM output. |
| -bed | When using BAM input (-abam), write output as BED. The default is to write output in BAM when using -abam. For example: bedtools window -abam reads.bam -b genes.bed -bed |
| -w | Base pairs added upstream and downstream of each entry in A when searching for overlaps in B. Default is 1000 bp. |
| -l | Base pairs added upstream (left of) of each entry in A when searching for overlaps in B. Allows one to create asymmetrical “windows”. Default is 1000bp. |
| -r | Base pairs added downstream (right of) of each entry in A when searching for overlaps in B. Allows one to create asymmetrical “windows”. Default is 1000bp. |
| -sw | Define -l and -r based on strand. For example if used, -l 500 for a negative-stranded feature will add 500 bp downstream. By default, this is disabled. |
| -sm | Only report hits in B that overlap A on the same strand. By default, overlaps are reported without respect to strand. |
| -Sm | Only report hits in B that overlap A on the opposite strand. By default, overlaps are reported without respect to strand. |
| -u | Write original A entry once if any overlaps found in B. In other words, just report the fact at least one overlap was found in B. |
| -c | For each entry in A, report the number of hits in B while restricting to -w, -l, and -r. Reports 0 for A entries that have no overlap with B. |
| -v | Only report those entries in A that have no overlaps with B. |
| -header | Print the header from the A file prior to results. |
Default behavior¶
By default, bedtools window adds 1000 bp upstream and downstream of each A
feature and searches for features in B that overlap this “window”. If an overlap
is found in B, both the original A feature and the original B feature are
reported.
$ cat A.bed
chr1 100 200
$ cat B.bed
chr1 500 1000
chr1 1300 2000
$ bedtools window -a A.bed -b B.bed
chr1 100 200 chr1 500 1000
-w Defining a custom window size¶
Instead of using the default window size of 1000bp, one can define a custom,
symmetric window around each feature in A using the -w option. One should
specify the window size in base pairs. For example, a window of 5kb should be
defined as -w 5000.
For example (note that in contrast to the default behavior, the second B entry is reported):
$ cat A.bed
chr1 100 200
$ cat B.bed
chr1 500 1000
chr1 1300 2000
$ bedtools window -a A.bed -b B.bed -w 5000
chr1 100 200 chr1 500 1000
chr1 100 200 chr1 1300 2000
-l and -r Defining asymmetric windows¶
One can also define asymmetric windows where a differing number of bases are
added upstream and downstream of each feature using the -l (upstream)
and -r (downstream)** options.
Note
By default, the -l and -r options ignore strand. If you want to define
upstream and downstream based on strand, use the -sw option (below)
with the -l and -r options.
For example (note the difference between -l 200 and -l 300):
$ cat A.bed
chr1 1000 2000
$ cat B.bed
chr1 500 800
chr1 10000 20000
$ bedtools window -a A.bed -b B.bed -l 200 -r 20000
chr1 1000 2000 chr1 10000 20000
$ bedtools window -a A.bed -b B.bed -l 300 -r 20000
chr1 1000 2000 chr1 500 800
chr1 1000 2000 chr1 10000 20000
-sw Defining asymmetric windows based on strand¶
Especially when dealing with gene annotations or RNA-seq experiments, you may
want to define asymmetric windows based on “strand”. For example, you may want
to screen for overlaps that occur within 5000 bp upstream of a gene (e.g. a
promoter region) while screening only 1000 bp downstream of the gene.
By enabling the -sw (“stranded” windows) option, the windows are added
upstream or downstream according to strand. For example, imagine one
specifies -l 5000, -r 1000 as well as the -sw option. In this case,
forward stranded (“+”) features will screen 5000 bp to the left (that is,
lower genomic coordinates) and 1000 bp to the right (that is, higher
genomic coordinates). By contrast, reverse stranded (“-”) features will screen
5000 bp to the right (that is, higher genomic coordinates) and 1000 bp to
the left (that is, lower genomic coordinates).
For example (note the difference between -l 200 and -l 300):
$ cat A.bed
chr1 10000 20000 A.forward 1 +
chr1 10000 20000 A.reverse 1 -
$ cat B.bed
chr1 1000 8000 B1
chr1 24000 32000 B2
$ bedtools window -a A.bed -b B.bed -l 5000 -r 1000 -sw
chr1 10000 20000 A.forward 1 + chr1 1000 8000 B1
chr1 10000 20000 A.reverse 1 - chr1 24000 32000 B2
-sm Enforcing matches with the same “strandedness”¶
This option behaves the same as the -s option for bedtools intersect
while scanning for overlaps within the “window” surrounding A. That is, overlaps
in B will only be included if the B interval is on the same strand as the A
interval.
-Sm Enforcing matches with the opposite “strandedness”¶
This option behaves the same as the -S option for bedtools intersect while
scanning for overlaps within the “window” surrounding A. That is, overlaps in
B will only be included if the B interval is on the opposite strand as the A
interval.
-u Reporting the presence/absence of at least one overlapping feature¶
This option behaves the same as for bedtools intersect. That is, even if
multiple overlaps exist, each A interval will only be reported once.
-c Reporting the number of overlapping features¶
This option behaves the same as for bedtools intersect. That is, it will
report the count of intervals in B that overlap each A interval.
-v Reporting the absence of any overlapping features¶
This option behaves the same as for bedtools intersect. That is, it will
only report those intervals in A that have have zero overlaps in B.
-header Print the header for the A file before reporting results.¶
By default, if your A file has a header, it is ignored when reporting results. This option will instead tell bedtools to first print the header for the A file prior to reporting results.
Example usage¶
Below are several examples of basic bedtools usage. Example BED files are provided in the /data directory of the bedtools distribution.
bedtools intersect¶
Report the base-pair overlap between sequence alignments and genes.
bedtools intersect -a reads.bed -b genes.bed
Report whether each alignment overlaps one or more genes. If not, the alignment is not reported.
bedtools intersect -a reads.bed -b genes.bed -u
Report those alignments that overlap NO genes. Like “grep -v”
bedtools intersect -a reads.bed -b genes.bed -v
Report the number of genes that each alignment overlaps.
bedtools intersect -a reads.bed -b genes.bed -c
Report the entire, original alignment entry for each overlap with a gene.
bedtools intersect -a reads.bed -b genes.bed -wa
Report the entire, original gene entry for each overlap with a gene.
bedtools intersect -a reads.bed -b genes.bed -wb
Report the entire, original alignment and gene entries for each overlap.
bedtools intersect -a reads.bed -b genes.bed -wa -wb
Only report an overlap with a repeat if it spans at least 50% of the exon.
bedtools intersect -a exons.bed -b repeatMasker.bed -f 0.50
Only report an overlap if comprises 50% of the structural variant and 50% of the segmental duplication. Thus, it is reciprocally at least a 50% overlap.
bedtools intersect -a SV.bed -b segmentalDups.bed -f 0.50 -r
Read BED A from stdin. For example, find genes that overlap LINEs but not SINEs.
bedtools intersect -a genes.bed -b LINES.bed | intersectBed -a stdin -b SINEs.bed -v
Retain only single-end BAM alignments that overlap exons.
bedtools intersect -abam reads.bam -b exons.bed > reads.touchingExons.bam
Retain only single-end BAM alignments that do not overlap simple sequence repeats.
bedtools intersect -abam reads.bam -b SSRs.bed -v > reads.noSSRs.bam
bedtools bamtobed¶
Convert BAM alignments to BED format.
bedtools bamtobed -i reads.bam > reads.bed
Convert BAM alignments to BED format using the BAM edit distance (NM) as the BED “score”.
bedtools bamtobed -i reads.bam -ed > reads.bed
Convert BAM alignments to BEDPE format.
bedtools bamtobed -i reads.bam -bedpe > reads.bedpe
bedtools window¶
Report all genes that are within 10000 bp upstream or downstream of CNVs.
bedtools window -a CNVs.bed -b genes.bed -w 10000
Report all genes that are within 10000 bp upstream or 5000 bp downstream of CNVs.
bedtools window -a CNVs.bed -b genes.bed -l 10000 -r 5000
Report all SNPs that are within 5000 bp upstream or 1000 bp downstream of genes. Define upstream and downstream based on strand.
bedtools window -a genes.bed -b snps.bed -l 5000 -r 1000 -sw
bedtools closest¶
Note: By default, if there is a tie for closest, all ties will be reported. closestBed allows overlapping features to be the closest.
Find the closest ALU to each gene.
bedtools closest -a genes.bed -b ALUs.bed
Find the closest ALU to each gene, choosing the first ALU in the file if there is a tie.
bedtools closest -a genes.bed -b ALUs.bed -t first
Find the closest ALU to each gene, choosing the last ALU in the file if there is a tie.
bedtools closest -a genes.bed -b ALUs.bed -t last
bedtools subtract¶
Note
If a feature in A is entirely “spanned” by any feature in B, it will not be reported.
Remove introns from gene features. Exons will (should) be reported.
bedtools subtract -a genes.bed -b introns.bed
bedtools merge¶
Note
merge requires that the input is sorted by chromosome and then by start
coordinate. For example, for BED files, one would first sort the input
as follows: sort -k1,1 -k2,2n input.bed > input.sorted.bed
Merge overlapping repetitive elements into a single entry.
bedtools merge -i repeatMasker.bed
Merge overlapping repetitive elements into a single entry, returning the number of entries merged.
bedtools merge -i repeatMasker.bed -n
Merge nearby (within 1000 bp) repetitive elements into a single entry.
bedtools merge -i repeatMasker.bed -d 1000
bedtools coverage¶
Compute the coverage of aligned sequences on 10 kilobase “windows” spanning the genome.
bedtools coverage -a reads.bed -b windows10kb.bed | head
chr1 0 10000 0 10000 0.00
chr1 10001 20000 33 10000 0.21
chr1 20001 30000 42 10000 0.29
chr1 30001 40000 71 10000 0.36
Compute the coverage of aligned sequences on 10 kilobase “windows” spanning the genome and created a BEDGRAPH of the number of aligned reads in each window for display on the UCSC browser.
bedtools coverage -a reads.bed -b windows10kb.bed | cut -f 1-4 > windows10kb.cov.bedg
Compute the coverage of aligned sequences on 10 kilobase “windows” spanning the genome and created a BEDGRAPH of the fraction of each window covered by at least one aligned read for display on the UCSC browser.
bedtools coverage -a reads.bed -b windows10kb.bed | \
awk '{OFS="\t"; print $1,$2,$3,$6}' \
> windows10kb.pctcov.bedg
bedtools complement¶
Report all intervals in the human genome that are not covered by repetitive elements.
bedtools complement -i repeatMasker.bed -g hg18.genome
bedtools shuffle¶
Randomly place all discovered variants in the genome. However, prevent them from being placed in know genome gaps.
bedtools shuffle -i variants.bed -g hg18.genome -excl genome_gaps.bed
Randomly place all discovered variants in the genome. However, prevent them from being placed in know genome gaps and require that the variants be randomly placed on the same chromosome.
bedtools shuffle -i variants.bed -g hg18.genome -excl genome_gaps.bed -chrom
Advanced usage¶
Mask all regions in a genome except for targeted capture regions.¶
Step 1. Add 500 bp up and downstream of each probe
bedtools slop -i probes.bed -g hg18.genome -b 500 > probes.500bp.bed
NB genome is two column chromosome size list - i.e. https://genome.ucsc.edu/goldenpath/help/hg18.chrom.sizes
Step 2. Get a BED file of all regions not covered by the probes (+500 bp up/down)
bedtools complement -i probes.500bp.bed -g hg18.genome > probes.500bp.complement.bed
Step 3. Create a masked genome where all bases are masked except for the probes +500bp
bedtools maskfasta -fi hg18.fa -bed probes.500bp.complement.bed \
-fo hg18.probecomplement.masked.fa
Screening for novel SNPs.¶
Find all SNPs that are not in dbSnp and not in the latest 1000 genomes calls
bedtools intersect -a snp.calls.bed -b dbSnp.bed -v | \
bedtools intersect -a - -b 1KG.bed -v | \
> snp.calls.novel.bed
Computing the coverage of features that align entirely within an interval.¶
By default, bedtools coverage counts any feature in A that overlaps B
by >= 1 bp. If you want to require that a feature align entirely within B for
it to be counted, you can first use intersectBed with the “-f 1.0” option.
bedtools intersect -a features.bed -b windows.bed -f 1.0 | \
bedtools coverage -a windows.bed -b - \
> windows.bed.coverage
Computing the coverage of BAM alignments on exons.¶
One can combine samtools with bedtools to compute coverage directly
from the BAM data by using bamtobed.
bedtools bamtobed -i reads.bam | \
bedtools coverage -a exons.bed -b - \
> exons.bed.coverage
Take it a step further and require that coverage be from properly-paired reads.
samtools view -uf 0x2 reads.bam | \
coverageBed -abam - -b exons.bed \
> exons.bed.proper.coverage
Computing coverage separately for each strand.¶
Use grep to only look at forward strand features (i.e. those that end in “+”).
bedtools bamtobed -i reads.bam | \
grep \+$ | \
bedtools coverage -a - -b genes.bed \
> genes.bed.forward.coverage
Use grep to only look at reverse strand features (i.e. those that end in “-“).
bedtools bamtobed -i reads.bam | \
grep \-$ | \
bedtools coverage -a - -b genes.bed \
> genes.bed.reverse.coverage
FAQ¶
Installation issues¶
Why am I getting all of these zlib errors?¶
On certain operating systems (especially free Linux distributions) the complete zlib libraries are not installed. Bedtools depends upon zlib in order to decompress gzipped files.
- Building main bedtools binary.
obj/gzstream.o: In function gzstreambuf::open(char const*, int):
gzstream.C:(.text+0x2a5): undefined reference to gzopen64'
collect2: ld returned 1 exit status
make: *** [all] Error 1
If you see an error such as the above, it suggests you need to install the
zlib and zlib1g-dev libraries. This is typically straightforward using
package managers. For example, on Debian/Ubuntu this would be:
apt-get install zlib
apt-get install zlib1g-dev
and on Fedora/Centos this would be:
yum install zlib
yum install zlib1g-dev
Compiling with a specific zlib library¶
If you need to override the location of the zlib library used to compile bedtools, you can run make and specify the LIBS argument. For example:
make LIBS='/PATH/TO/ZLIB/lib/libz.a'
General questions¶
How do I know what version of bedtools I am using?¶
Use the –version option.
$ bedtools --version
bedtools v2.17.0
Issues with output¶
I know there are overlaps, but none are reported. What might be wrong?¶
There are two common causes of this problem. The first cause is non-obvious
differences in the way chromosomes are named in files being compared.
For example, “1” is not the same as “chr1” just as ” chr1” is not the same
as “chr1”. Secondly, users often copy files from a Windows machine to a UNIX
machine. This causes issues because Windows uses two bytes to represent
the end of a line (\r\n) whereas the UNIX convention uses a single byte
(\n). If your files don’t conform to the UNIX convention, you will have
problems. One can convert files from Windows to UNIX with
the following command:
perl -i -p -e 's/\r\n/\n/g;' file.windows > file.unix
Installation issues¶
Related software¶
Bedtools has been used as an engine behind other genomics software and has been integrated into widely used tools such as Galaxy and IGV. Below is a likely incomplete list. If you know of others, please let us know, or better yet, edit the document on GitHub and send us a pull request. You can do this by clicking on the “Edit and improve this document” link in the lower lefthand corner.
IGV¶
Bedtools is now integrated into the IGV genome viewer as of IGV version 2.2. We are actively working withe IGV development team to improve and expand this integration. See here and here for details.
Galaxy¶
Galaxy has its own tools for working with genomic intervals under the “Operate on Genomic Intervals” section. A subset of complementary Bedtools utilities have also been made available on Galaxy in an effort to provide functionality that isn’t available with the native Galaxy tools.

Pybedtools¶
Pybedtools is a really fantastic Python library that wraps (and extends upon) the bedtools utilities and exposes them for easy use and new tool development using Python. Pybedtools is actively maintained by Ryan Dale.
MISO¶
MISO is “a probabilistic framework
that quantitates the expression level of alternatively spliced genes from
RNA-Seq data, and identifies differentially regulated isoforms or exons across
samples.” A subset of the functionality in MISO depends upon bedtools. MISO
is developed by Yarden Katz.
RetroSeq¶
RetroSeq is “a tool for discovery and genotyping of transposable element variants (TEVs) (also known as mobile element insertions) from next-gen sequencing reads aligned to a reference genome in BAM format”. RetroSeq is developed by Thomas Keane. Source code can be obtained on GitHub.
Intersphinx documentation¶
BEDTools documentation pages are available via Intersphinx (http://sphinx-doc.org/ext/intersphinx.html). To enable this, add the following to conf.py in a Sphinx project:
intersphinx_mapping = {‘bedtools’: (’http://bedtools.readthedocs.org/en/latest/’, None)}
BEDtools documentation links can then be generated with e.g.:
Performance¶
As of version 2.18, bedtools is substantially more scalable thanks to improvements we have made in the algorithm used to process datasets that are pre-sorted
by chromosome and start position. As you can see in the plots below, the speed and memory consumption scale nicely
with sorted data as compared to the poor scaling for unsorted data. The current version of bedtools intersect is as fast as (or slightly faster) than the bedops package’s bedmap which uses a similar algorithm for sorted data. The plots below represent counting the number of intersecting alignments from exome capture BAM files against CCDS exons.
The alignments have been converted to BED to facilitate comparisons to bedops. We compare to the bedmap --ec option because similar error checking is enforced by bedtools.
Note: bedtools could not complete when using 100 million alignments and the R-Tree algorithm used for unsorted data owing to a lack of memory.
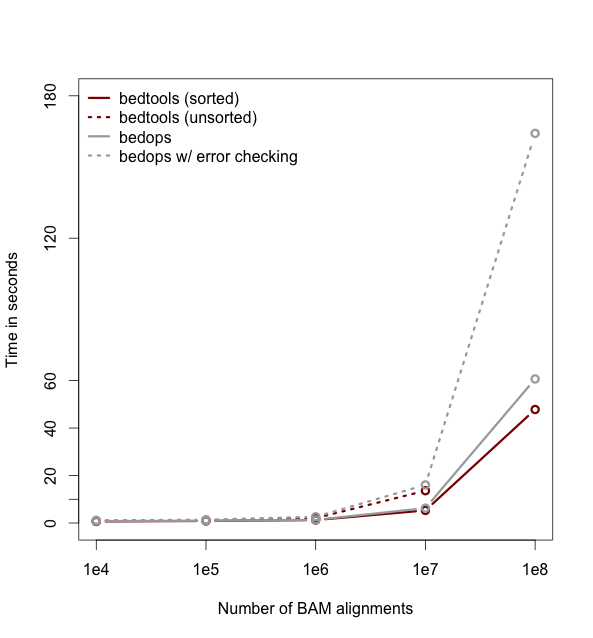
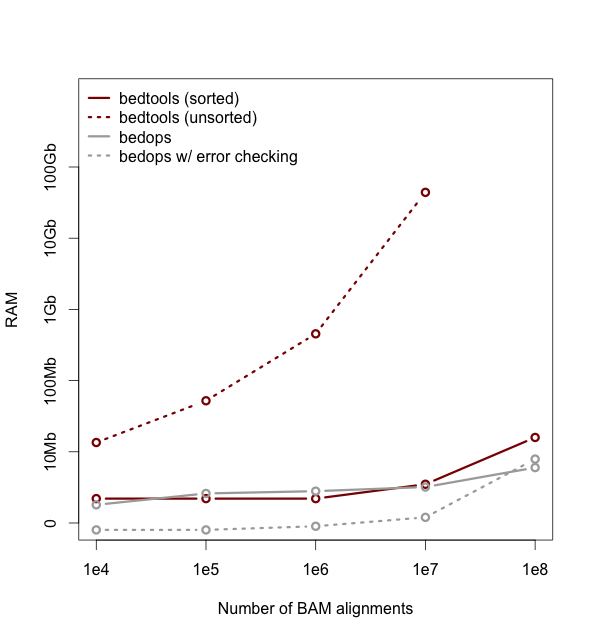
Commands used:
# bedtools sorted
$ bedtools intersect \
-a ccds.exons.bed -b aln.bam.bed \
-c \
-sorted
# bedtools unsorted
$ bedtools intersect \
-a ccds.exons.bed -b aln.bam.bed \
-c
# bedmap (without error checking)
$ bedmap --echo --count --bp-ovr 1 \
ccds.exons.bed aln.bam.bed
# bedmap (no error checking)
$ bedmap --ec --echo --count --bp-ovr 1 \
ccds.exons.bed aln.bam.bed
Brief example¶
Let’s imagine you have a BED file of ChiP-seq peaks from two different experiments. You want to identify peaks that were observed in both experiments (requiring 50% reciprocal overlap) and for those peaks, you want to find to find the closest, non-overlapping gene. Such an analysis could be conducted with two, relatively simple bedtools commands.
# intersect the peaks from both experiments.
# -f 0.50 combined with -r requires 50% reciprocal overlap between the
# peaks from each experiment.
$ bedtools intersect -a exp1.bed -b exp2.bed -f 0.50 -r > both.bed
# find the closest, non-overlapping gene for each interval where
# both experiments had a peak
# -io ignores overlapping intervals and returns only the closest,
# non-overlapping interval (in this case, genes)
$ bedtools closest -a both.bed -b genes.bed -io > both.nearest.genes.txt
License¶
bedtools is freely available under a GNU Public License (Version 2).
Acknowledgments¶
To do.
Mailing list¶
If you have questions, requests, or bugs to report, please email the bedtools mailing list

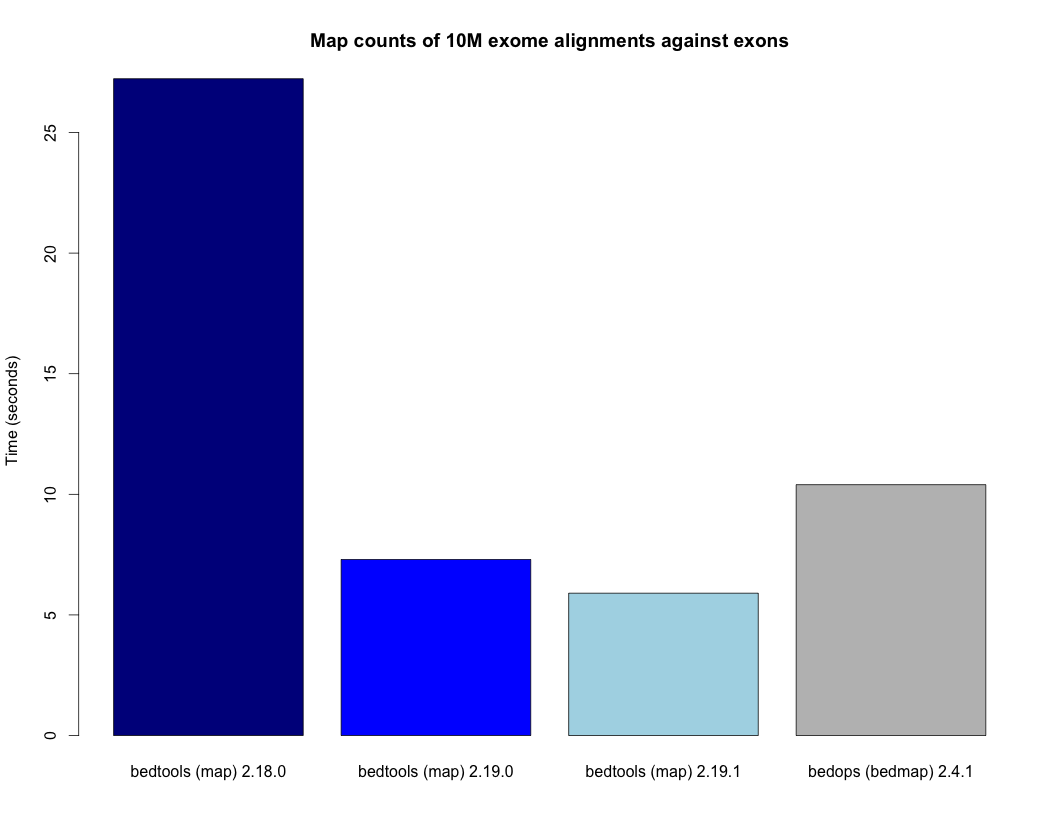{kind=link}
{kind=link}